Article Text

Abstract
Five cases of the Hirschsprung’s disease–congenital central hypoventilation syndrome (CCHS) association are presented and 41 other published cases reviewed. These children have a distinct pattern of associated features, an equal sex incidence, and a characteristic spectrum of disease severity which suggests that the condition is genetically distinct from other cases of Hirschsprung’s disease. While approximately 1.5% of Hirschsprung’s disease patients, and 10% of those with total colonic aganglionosis, will have CCHS, up to 50% of CCHS patients will have Hirschsprung’s disease. Approximately 20% of CCHS/Hirschsprung patients will also have neuroblastoma or ganglioneuroma, usually multiple. Abnormalities of the eye and autonomic nervous system are also common. The ventilatory abnormality is usually evident on the first day of life. The aganglionosis is also severe, with more than half (59%) of the patients having aganglionosis extending into the small bowel.
- Hirschsprung’s disease
- congenital central hypoventilation syndrome
- total colonic aganglionosis
- neurocristopathy
Statistics from Altmetric.com
- Hirschsprung’s disease
- congenital central hypoventilation syndrome
- total colonic aganglionosis
- neurocristopathy
Hirschsprung’s disease is a condition caused by congenital absence of ganglion cells from the enteric nervous system, resulting in bowel obstruction ranging in severity from chronic severe constipation to complete obstruction and early neonatal death. It is thought to originate in a failure of migration of neural crest derived precursor cells,1 although this is controversial and a hostile gut microenvironment may also contribute. Congenital central hypoventilation syndrome (CCHS) results in hypoventilation, most pronounced during sleep, with relative insensitivity to hypercarbia and a lesser insensitivity to hypoxia, in the absence of other abnormalities of the cardiorespiratory system. CCHS is also known as Ondine’s curse, after a figure from Germanic mythology. Ondine’s curse properly refers only to the condition where hypoventilation is restricted to the sleeping state. CCHS and Hirschsprung’s disease were first reported together by Haddad et al in 1978.2 Both are uncommon, and their co-occurrence suggests a common aetiology, probably involving a fault of neural crest development.
We here report the largest single series (five cases) of the congenital central hypoventilation syndrome–Hirschsprung’s disease association (“Haddad syndrome”). These children have presented to the children’s hospitals of New South Wales and the Australian Capital Territory over the past 21 years, adding to the 41 already reported in English language journals. These children present a challenging spectrum of problems and represent a subgroup of more severely affected children with Hirschsprung’s disease.
Methods
We identified cases by a retrospective review of Hirschsprung’s disease admissions to children’s hospitals in New South Wales and the Australian Capital Territory over the 21 year period from 1 January 1975 to 1 January 1996. During this period there were 341 admissions with this diagnosis to the three teaching hospitals. Patients who had the concomitant diagnosis of CCHS were then reviewed separately. At the time of writing two children are known to have died. The families of surviving children were contacted for follow up interview and the collection of blood for genetic studies.
We also reviewed published reports. Forty one previously reported cases of the association are summarised in table 1. An additional 10 patients have been reported without supporting detail25 26; these are not included in the analysis.
Summary of clinical details of 41 cases of Hirschsprung’s disease–congenital hypoventilation syndrome association
Results
Clinical details of our five patients are summarised in tables 2and 3. All had biopsy proved Hirschsprung’s disease, and evidence of congenital central hypoventilation on monitoring in the intensive care unit or with formal sleep studies. There are two males and three females. Treatment was withdrawn from two, and there are three survivors. These children are now satisfactorily managed on home ventilation.
Clinical features of CCHS/Hirschsprung patients
Associated features in CCHS/Hirschsprung patients
Three children had sigmoid aganglionosis and two had total colonic aganglionosis. In three patients this was confirmed on histology, and in two patients (one from each group) in whom the parents refused necropsy it was inferred from barium studies and rectal biopsy. Although barium enema is known to be misleading in as many as 30% of cases,27 the presence of numerous nerve twigs in the rectal suction biopsy suggests less than total colonic aganglionosis.
Two of our cases had gastro-oesophageal reflux, one had dyscoordinated swallowing on barium swallow, and one had a complete absence of swallowing reflexes. Information on the fifth case is inconclusive.
In this series there was no definitive evidence of neural crest tumour, although one case had undiagnosed retroperitoneal masses at the time of death. Features of autonomic dysfunction are common in other reports, and have been noted in table 1. An abnormal facies was noted in one case. The two surviving females have both had karyotype examinations, which are normal female to the 450 and 500 band stages of resolution, respectively. The surviving male is overseas and therefore unavailable for study at the time of writing.
Discussion
Congenital central hypoventilation syndrome results in a decrease in the depth rather than the rate of breathing, most severe during quiet sleep and less marked in rapid eye movement sleep. There is, however, a spectrum of severity, so that the more severely affected patients hypoventilate when both asleep and awake, and rate may also be affected. Insensitivity to hypercapnia is the most constant finding, with a variable response to hypoxaemia.28
A recent report suggested that up to 30% of patients with Hirschsprung’s disease may have some form of dysautonomia (Staiano A,et al: Autonomic dysfunction in children with Hirschsprung’s disease. Abstract from 29th Annual Meeting of European Society for Gastroenterology and Nutrition, Munich, 5–8 June 1996). If this observation is true, then the CCHS/Hirschsprung combination may well represent the severe end of the spectrum for autonomic manifestations as well as for length of aganglionosis.
SEX RATIO AND LENGTH OF AGANGLIONOSIS
Of the 46 reported cases (including our own five), 21 were female and the sex of three was not stated. This 1:1 ratio is consistent with previous observations that the sex ratio in Hirschsprung’s disease, which normally has a 4:1 male preponderance, approaches unity as the length of the involved segment increases.29 The length of aganglionosis was definitely known for 34 of these cases, with another six doubtful and six unknown. There was no definite evidence that females were affected more or less severely than males; however, equal numbers of males and females (eight each) had aganglionosis affecting the whole colon or ileum. The four most severely affected patients—with aganglionosis extending into the mid-small bowel or more proximally—were all boys. Counting three whose sex was unknown, 24 of the 40 in whom the information was available (60%) had aganglionosis that probably (n = 3) or certainly (n = 21) affected at least the whole colon.
Of those with less than total colonic aganglionosis, five of 16 had long segment Hirschsprung’s disease with aganglionosis to the splenic flexure or beyond. Only eight had aganglionosis of sigmoid length or less, a striking reversal of the usual length distribution in Hirschsprung’s disease (fig 1; table 4). Although the length distribution remains bimodal, the major peak of the distribution in CCHS/Hirschsprung patients is at the level of the ileocaecal valve, and there is a gradual tapering of the distribution into the proximal bowel. The difference of the distribution in CCHS/Hirschsprung patients compared with both the Down’s syndrome/Hirschsprung combination and non-syndromic Hirschsprung’s disease suggests a different aetiological mechanism for the former. The bimodal distribution implies that migrating ganglion cells may have to cross potential barriers at the level of ileocaecal valve and rectosigmoid junction, which become impassable in the disease state. Alternatively, it might suggest that the bowel is populated by a small number of discrete clones and their progeny: if one clone is missing, sigmoid aganglionosis results; if several are missing, total colonic aganglionosis occurs (Cass DT: Abstract from First International Workshop on Hirschsprung’s Disease, Genoa, Italy, March 1994).
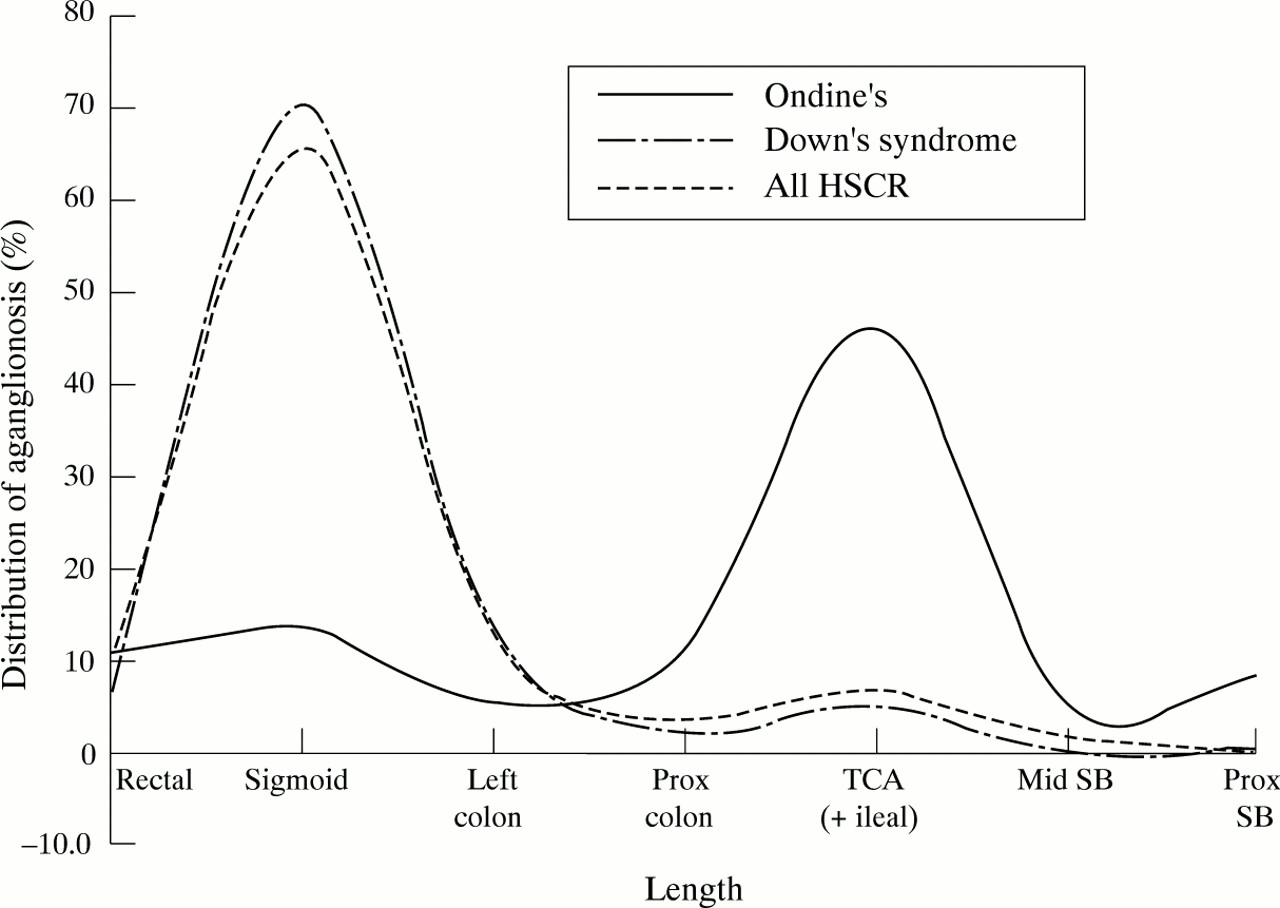
{kind=link}
Distribution of aganglionosis in cases of Hirschsprung’s disease (HSCR). SB=small bowel; TCA=total colonic aganglionosis.
Length of aganglionosis. Comparing lengths of aganglionosis between collected CCHS/Hirschprung cases, and the Sydney 30 year experience of Hirschprung’s disease alone, or with Down’s syndrome
Recent work has shown that the penetrance of mutations in the Hirschsprung’s disease associated genes RET and the endothelin B receptor (ENDRB) is affected by sex.30 31 The equal sex incidence in CCHS/Hirschsprung patients suggests that a different mechanism may be at work in this condition. The very high incidence of associated autonomic anomalies supports this possibility by suggesting that the underlying anomaly may operate at an earlier stage of development than RET or ENDRB mutations, or over a longer segment of neural crest.
PATTERNS OF HYPOVENTILATION
While there are few data on normal respiratory values in infancy,32 of the patients with CCHS alone at our institution, two on home ventilation both presented late—one at 12 months, and one at 6 years (Seton C, personal communication). A third patient (who has since died) presented at the age of 3 years. In clear contrast to this, 90% of the cases in this collected series of CCHS/Hirschsprung patients were ventilated on day 1 of life: the median age for starting ventilation in the 25 patients for whom the data are available was 3 hours. Khalifa et al asserted in 1988 that most cases of primary central hypoventilation occur in adult life, and that CCHS may represent a more severe form of this disease.33 A recent paper noted that there was an excess of sudden infant death syndrome (SIDS) in relatives of patients with CCHS.23 If SIDS is part of the same disease spectrum as CCHS, then clearly the CCHS/Hirschsprung combination represents another variant, as it is also at the more severe end of the Hirschsprung’s disease spectrum.
ASSOCIATED FEATURES
Eyes
Overall 10 of the 46 patients reviewed here (21.7%) were noted to have abnormalities of the pupils, extraocular muscles, or eyelids, either unilaterally or bilaterally. In a series of 32 patients with CCHS (with or without Hirschsprung’s disease) reviewed by Weese-Mayeret al, as many as 60% were said to have ophthalmic abnormalities.28 Three of the five patients newly reported here have ophthalmic abnormalities, consistent with Weese-Mayer’s report; this suggests that the low incidence in the collected series probably represents underreporting rather than a true difference in incidence.
Swallowing
An abnormality of swallowing or oesophageal motility was identified in four of our five patients. This suggests a generalised abnormality of gastrointestinal motility. This tendency is confirmed in the broader series, where overall 10 of 46 had evidence of oesophageal dysmotility, with or without documented reflux.
Hearing
Abnormal brain stem auditory evoked responses were identified in five of the 46 patients. It is not possible to say what percentage of CCHS/Hirschsprung children have a sensorineural hearing deficit as it is uncertain how often this was tested for. However, sensorineural deafness forms part of the Shah-Waardenburg syndrome, now known to be related to a defect in the endothelin signalling pathway.34 Two of three children with near total intestinal aganglionosis but without CCHS who had brain stem auditory evoked response testing showed abnormalities consistent with inner ear dysfunction.35 These results suggest that hearing loss in children with very long segment aganglionosis is likely to be more common than previously believed and should be tested for.
Neuroblastoma
There have been several previous reports of the association of CCHS and Hirschsprung’s disease with neuroblastoma, and neuroblastoma with either of these conditions on its own.36-38
Of the 46 cases reviewed here, there were seven (15.2%) with confirmed neuroblastoma or ganglioneuroma. Our patient with perirenal masses on ultrasound would make an eighth. Thus 17.4% of the total group have sympathetic chain tumours. These tumours tend to be multiple and bilateral when present. There is no evidence that they were more common in the children with longer segment disease in this series. Three further patients with Haddad syndrome were reported to have raised catecholamines on at least one occasion, but without other evidence of neuroblastoma.22 Unfortunately, the falling rate of necropsy examinations makes it hard to be accurate in estimating the real incidence of neuroblastoma and related tumours in these children. Neuroblastoma should, however, be looked for in all children with the Haddad syndrome. This significant association suggests that adrenal tissue from these children is worthy of further study.
Other conditions
Various other conditions reported in association with this syndrome are listed in table 1. Three patients had facial dysmorphism, but so far there is no characteristic facial feature that will identify these children. Hypotonia and fits have often been reported (in 13 of the 46 patients); although these may be secondary to hypoxia, they could also represent a primary phenomenon. The mother of one of our patients who required antiepileptic treatment for several years also has a history of seizure disorder, pointing to the possibility of an inherited tendency.
INCIDENCE
The five cases we report here were identified by retrospective review of cases of Hirschsprung’s disease presenting to New South Wales and the Australian Capital Territory teaching hospitals over 21 years. The total number of cases of Hirschsprung’s disease presenting to these hospitals in the same period was 341. On this basis, the CCHS/Hirschsprung combination accounts for 1.5% of all Hirschsprung’s disease cases. Uncomplicated CCHS presents to our sleep unit at a rate of much less than one case a year, and there are only two patients currently on long term home ventilation in the study area for pure CCHS without Hirschsprung’s disease. A third patient has recently died (Seton C, Rosier M; personal communication). Currently there are three living CCHS/Hirschsprung patients from this series. CCHS/Hirschsprung’s disease therefore comprises 50% of our CCHS population at most. In a survey of 32 CCHS patients in 1992, Weese Mayer et al found five patients (16%) with associated Hirschsprung’s disease.28 In a subsequent paper the same investigators stated that there are fewer than 100 published cases of CCHS (Weese-Mayer DE, Silvestri JM, Kenny AS, et al: Characterisation of CCHS. Paper presented at the Second International Hirschsprung disease meeting, Cleveland, Ohio, October 1995). The fact that our review now extends the number of cases of the CCHS/Hirschsprung association to 46 suggests that the risk of Hirschsprung’s disease for CCHS patients may be higher than 16% (although it is possible that the CCHS/Hirschsprung syndrome may be relatively overreported compared with CCHS alone). El-Halaby and Coran found that three of their seven CCHS patients on home ventilation (43%) had coexistent Hirschsprung’s disease,18 a figure that broadly agrees with our own figure of 50%.
GENETICS
A family history was recorded in only three of our patients, and in none of these was there a family history of Hirschsprung’s disease or CCHS. The family history is unknown or not stated in 21 of the previous 41 cases, but there are two sibling pairs in the 18 previous families for whom a family history is known, suggesting that the recurrence risk is similar to the recurrence risk for total colonic Hirschsprung’s disease (approximately 10%). Of our three families for which we do have a family history, two have a history of thyroid disease, and one of pigmentary anomalies. These features may both be linked to separate specific Hirschsprung’s disease related genes.34 39
A familial occurrence has been reported for both Hirschsprung’s disease and CCHS separately. Complex segregation analysis suggests that the inheritance of CCHS is the same with or without Hirschsprung’s disease, although this is based on only one positive sibship out of 50 CCHS probands.25
It is of interest that Haddad syndrome has been transmitted as an apparently autosomal dominant characteristic to two half sisters with a common father.3 However, no defects in any of the known Hirschsprung’s disease related genes have yet been reported in the CCHS/Hirschsprung association. Screening of the RET proto-oncogene has failed to show any mutation in Hirschsprung’s disease or CCHS patients,4 and only one case report of a point mutation in the endothelin 3 gene in a case of pure CCHS has been published.5 Involvement of the endothelin signalling pathway may account for the frequent observation of abnormal brain stem auditory evoked potentials in these children, as mutations in endothelin 3 and the endothelin B receptor (ENDRB) are known to be associated with sensorineural deafness and Hirschsprung’s disease. Interestingly, a point mutation in this gene in the Mennonite kindred is associated with a shorter segment form of Hirschsprung’s disease, deafness, and pigmentary anomalies, unlike the long segment disease seen in CCHS/Hirschsprung patients.30 Edery et al have reported a case of total colonic aganglionosis associated with deafness and depigmentation in an inbred family carrying an endothelin 3 mutation.40 If the endothelin pathway is involved in this condition, it seems likely that modifiers also exist. If EDNRB and RET are not involved, then the RET ligand, glial derived neurotrophic factor (GDNF), remains a candidate gene. We have screened two of our five patients for GDNF mutations, but have found no evidence of mutations in this gene in these patients.
TREATMENT
The combination of a condition requiring long term ventilation with long segment Hirschsprung’s disease, and its attendant problems, is a daunting one; however, three of our five patients remain alive and in good health at the time of most recent follow up. The two children who died in our series did so after a considered withdrawal of treatment.
There have been 19 deaths from among the 41 patients in this series in whom the outcome is known. Six of those who died have been reported since 1990. They have a mean survival of 19 months. Those reported before 1990 (13 cases) died at an average age of 5.9 months. It is not surprising that improvements in total parenteral nutrition and home ventilatory support should be reflected in improved survival, and the improvement parallels the better prognosis of other children with total colonic aganglionosis over the last 20 years. Anecdotal reports (cited by Weese-Mayer at the International Hirschsprung’s disease meeting in Cleveland in 1995) suggest that the first survivors of CCHS are now reaching adulthood, and that they have the potential for a healthy and largely normal existence, at least into their twenties.
Because of the well recognised link between total colonic aganglionosis and CCHS we suggest that all children with the former should undergo sleep monitoring after diagnosis, to rule out milder or less obvious forms of CCHS. It is possible that routine monitoring of other Hirschsprung’s disease patients would pick up subtle and previously unrecognised ventilatory changes. In fact two of 19 children with total colonic aganglionosis treated in New South Wales since 1 January 1975 have clinical CCHS. Thus CCHS must be regarded as a significant risk in children with total colonic aganglionosis. Similarly, clinicians who make the diagnosis of CCHS should be aware that Hirschsprung’s disease is common in these children, and a rectal suction biopsy should be considered if there is any concern about gut function. Finally, because of the high incidence of associated conditions, these children should be carefully assessed by their attending surgeon and physician, with particular care to exclude neural crest tumours and sensorineural deafness.
SUMMARY
The combination of CCHS and Hirschsprung’s disease is a condition distinct from classical Hirschsprung’s disease, and of greater severity. It occurs in approximately 1.5% of all cases of Hirschsprung’s disease, and in 10% of cases of total colonic aganglionosis. The CCHS/Hirschsprung combination contributes up to half the case load of CCHS referred to respiratory units for home ventilation. Infants presenting to neonatal units with either total colonic aganglionosis or CCHS alone should be screened for the other condition. Of all the children reviewed, approximately half are dead, half are female, and half have total colonic Hirschsprung’s disease.
There is a high risk of associated neuroblastoma, which may occur in nearly 20% of cases. This should be looked for whenever the CCHS/Hirschsprung combination is seen. Other associated problems are common; while these may be less severe they are clinically important, and include sensorineural deafness, ocular problems, and disturbances of gastrointestinal motility.
Management is difficult, and a multidisciplinary approach in a well supported referral centre is ideal. Although we can afford to be guardedly optimistic in prognosis, even in 1997 some families will opt to withdraw treatment, and this wish should be respected.
Acknowledgments
We wish to thank the Royal Australasian College of Surgeons, which gave us a grant that partly supported this work. We extend our thanks also to Dr C Seton and Dr K Waters of the Royal Alexandra Hospital sleep unit for their comments and advice.