First Identification of RNA-Binding Proteins That Regulate Alternative Exons in the Dystrophin Gene

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Selection of the Candidate RBPs
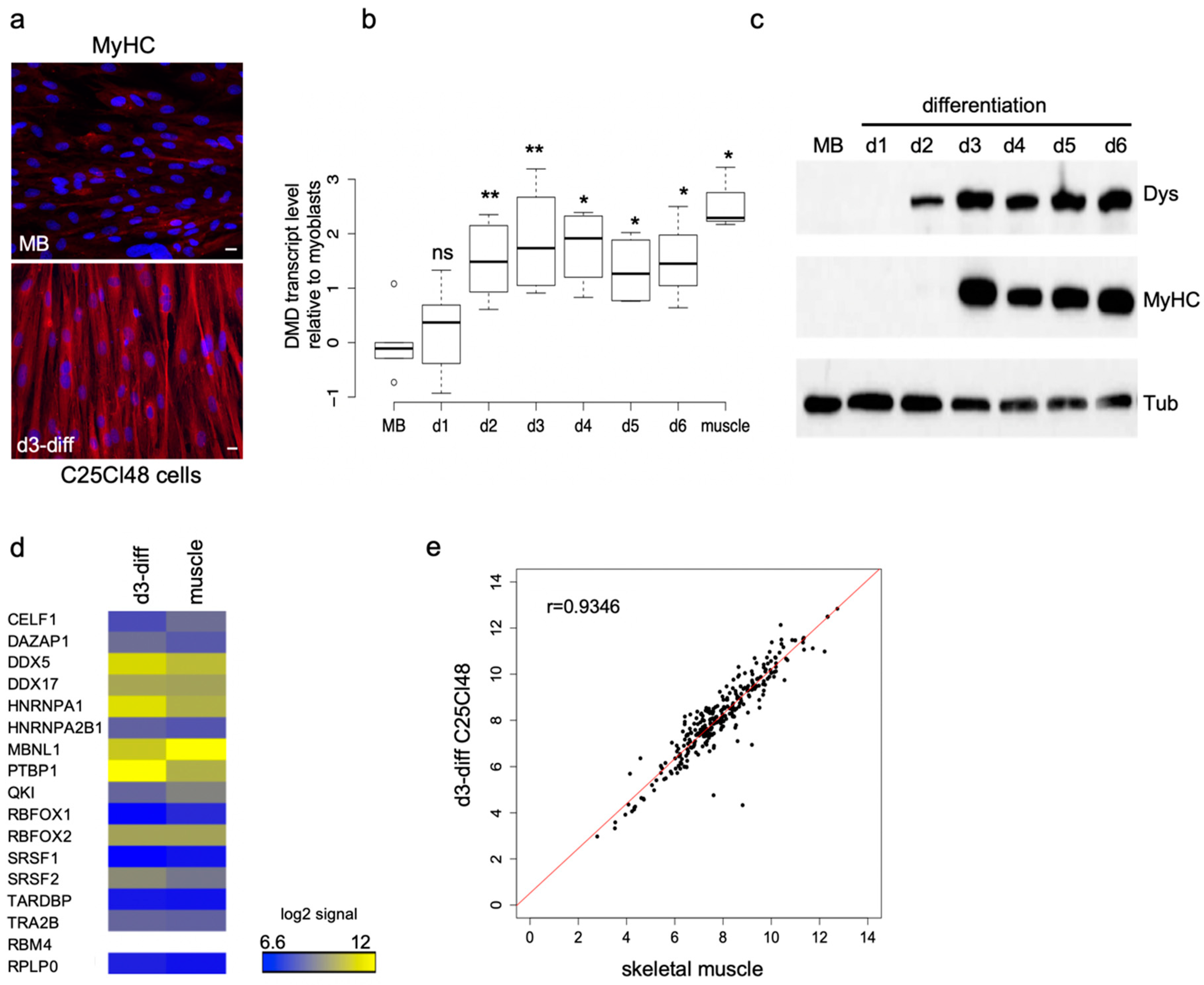
2.2. Characteristics of the Human Muscular C25Cl48 Cell Line
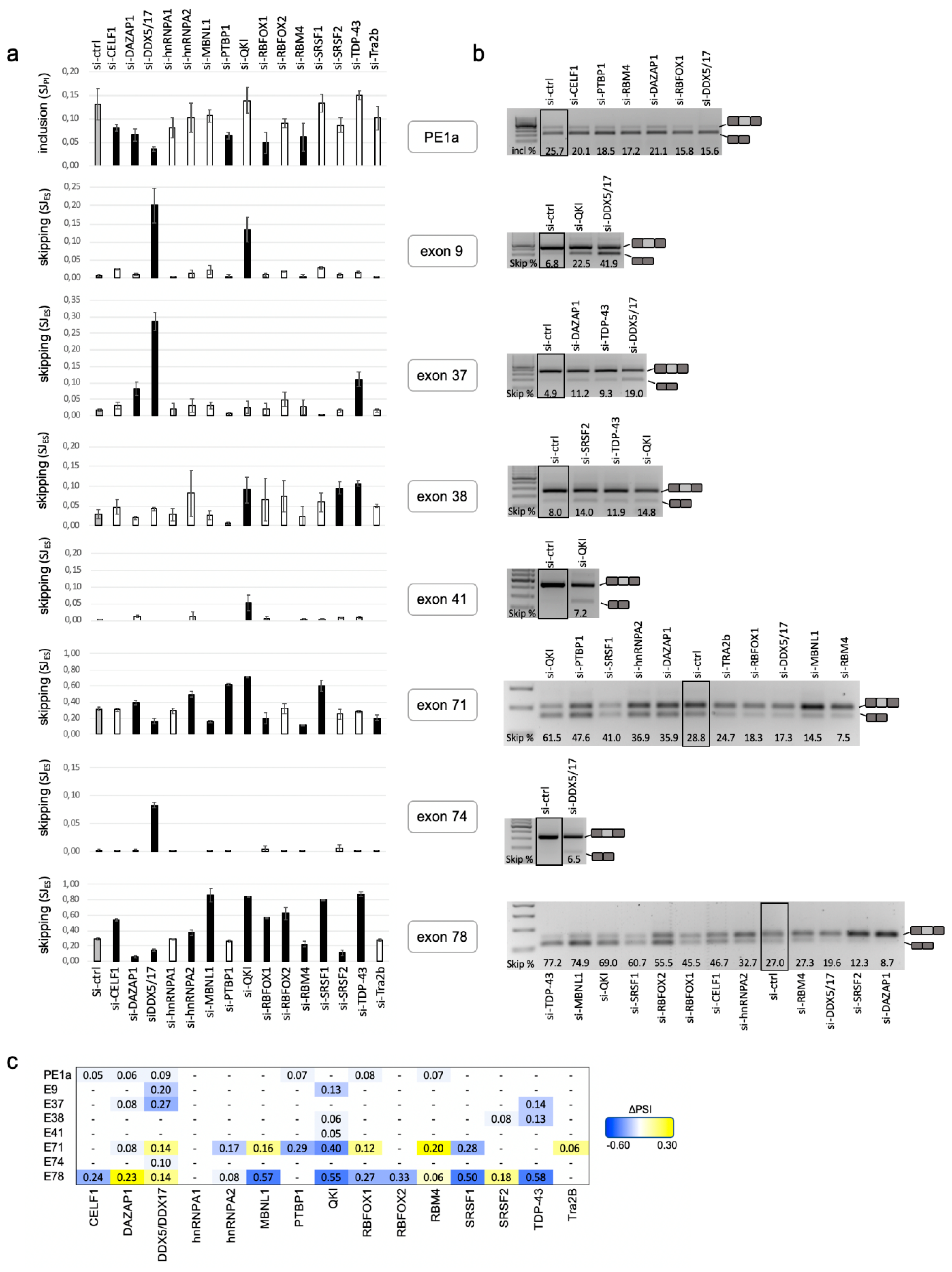
2.3. A Set of DMD Exons Is Sensitive to Changes in Cellular Abundance of RBPs
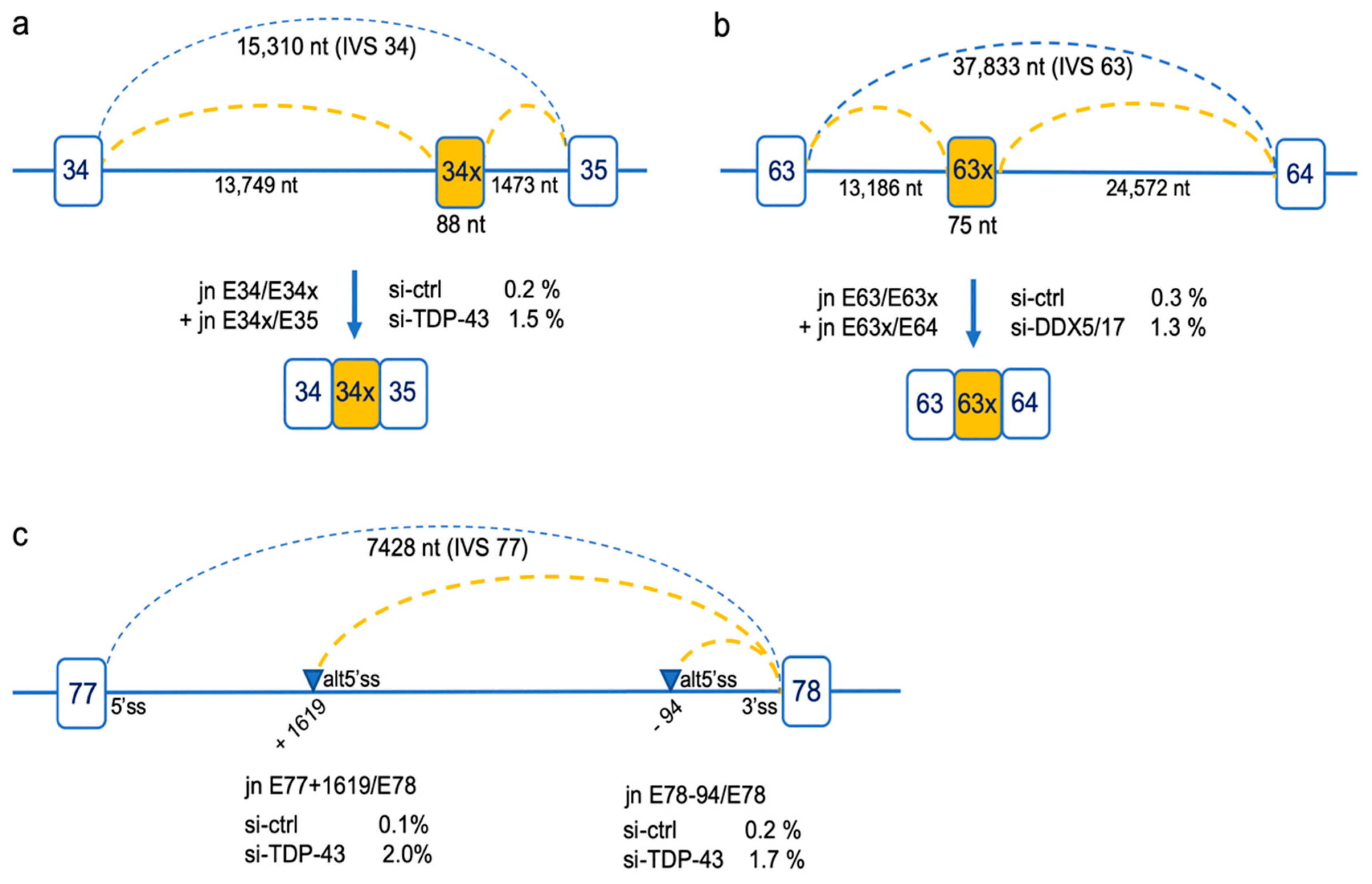
2.4. Rare Splicing Events Revealing Differentially Used Deep Intronic Splice Sites
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. siRNA-Mediated Depletion of RBPs
4.3. DMD-Targeted RNA-Seq
4.3.1. Sample Preparation
4.3.2. Long Range-Polymerase Chain Reaction (LR-PCR) and 454-Roche Library Construction and Sequencing
4.3.3. Bioinformatics Analysis
4.3.4. Availability of Data
4.4. RT-PCR Validation and RT-qPCR
4.5. Western Blotting
4.6. Microarray Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Comp. Physiol. 2015, 5, 1223–1239. [Google Scholar]
- Adams, M.E.; Odom, G.L.; Kim, M.J.; Chamberlain, J.S.; Froehner, S.C. Syntrophin binds directly to multiple spectrin-like repeats in dystrophin and mediates binding of nNOS to repeats 16-17. Hum. Mol. Genet. 2018, 27, 2978–2985. [Google Scholar] [CrossRef] [PubMed]
- Norwood, F.L.; Harling, C.; Chinnery, P.F.; Eagle, M.; Bushby, K.; Straub, V. Prevalence of genetic muscle disease in Northern England: In-depth analysis of a muscle clinic population. Brain 2009, 132, 3175–3186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Tyers, L.; Davids, L.M.; Wilmshurst, J.M.; Esterhuizen, A.I. Skin cells for use in an alternate diagnostic method for Duchenne muscular dystrophy. Neuromuscul. Disord. 2018, 28, 553–563. [Google Scholar] [CrossRef]
- Lederfein, D.; Levy, Z.; Augier, N.; Mornet, D.; Morris, G.; Fuchs, O.; Yaffe, D.; Nudel, U. A 71-kilodalton protein is a major product of the Duchenne muscular dystrophy gene in brain and other nonmuscle tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 5346–5350. [Google Scholar] [CrossRef] [Green Version]
- Bar, S.; Barnea, E.; Levy, Z.; Neuman, S.; Yaffe, D.; Nudel, U. A novel product of the Duchenne muscular dystrophy gene which greatly differs from the known isoforms in its structure and tissue distribution. Biochem. J. 1990, 272, 557–560. [Google Scholar] [CrossRef]
- Barbosa-Morais, N.L.; Irimia, M.; Pan, Q.; Xiong, H.Y.; Gueroussov, S.; Lee, L.J.; Slobodeniuc, V.; Kutter, C.; Watt, S.; Colak, R.; et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012, 338, 1587–1593. [Google Scholar] [CrossRef] [Green Version]
- Merkin, J.; Russell, C.; Chen, P.; Burge, C.B. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science 2012, 338, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Raj, B.; Blencowe, B.J. Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 2015, 87, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Lidov, H.G.; Kunkel, L.M. Dp140: Alternatively spliced isoforms in brain and kidney. Genomics 1997, 45, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Tadayoni, R.; Rendon, A.; Soria-Jasso, L.E.; Cisneros, B. Dystrophin Dp71: The smallest but multifunctional product of the Duchenne muscular dystrophy gene. Mol. Neurobiol. 2012, 45, 43–60. [Google Scholar] [CrossRef]
- Nishida, A.; Minegishi, M.; Takeuchi, A.; Awano, H.; Niba, E.T.; Matsuo, M. Neuronal SH-SY5Y cells use the C-dystrophin promoter coupled with exon 78 skipping and display multiple patterns of alternative splicing including two intronic insertion events. Hum. Gene. 2015, 134, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Aragón, J.; González-Reyes, M.; Romo-Yáñez, J.; Vacca, O.; Aguilar-González, G.; Rendón, A.; Vaillend, C.; Montañez, C. Dystrophin Dp71 Isoforms Are Differentially Expressed in the Mouse Brain and Retina: Report of New Alternative Splicing and a Novel Nomenclature for Dp71 Isoforms. Mol. Neurobiol. 2018, 55, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Bougé, A.L.; Murauer, E.; Beyne, E.; Miro, J.; Varilh, J.; Taulan, M.; Koenig, M.; Claustres, M.; Tuffery-Giraud, S. Targeted RNA-Seq profiling of splicing pattern in the DMD gene: Exons are mostly constitutively spliced in human skeletal muscle. Sci. Rep. 2017, 7, 39094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feener, C.A.; Koenig, M.; Kunkel, L.M. Alternative splicing of human dystrophin mRNA generates isoforms at the carboxy terminus. Nature 1989, 338, 509–511. [Google Scholar] [CrossRef]
- Rau, F.; Lainé, J.; Ramanoudjame, L.; Ferry, A.; Arandel, L.; Delalande, O.; Jollet, A.; Dingli, F.; Lee, K.Y.; Peccate, C.; et al. Abnormal splicing switch of DMD’s penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat. Comm. 2015, 6, 7205. [Google Scholar] [CrossRef] [Green Version]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat.Rev. Mol. Cell. Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Weyn-Vanhentenryck, S.M.; Feng, H.; Ustianenko, D.; Duffié, R.; Yan, Q.; Jacko, M.; Martinez, J.C.; Goodwin, M.; Zhang, X.; Hengst, U.; et al. Precise temporal regulation of alternative splicing during neural development. Nat. Commun. 2018, 9, 2189. [Google Scholar] [CrossRef] [Green Version]
- Witten, J.T.; Ule, J. Understanding splicing regulation through RNA splicing maps. Trends Genet. 2011, 27, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Hinkle, E.R.; Wiedner, H.J.; Black, A.J.; Giudice, J. RNA processing in skeletal muscle biology and disease. Transcription 2019, 10, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Bland, C.S.; Wang, E.T.; Vu, A.; David, M.P.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res. 2010, 38, 7651–7664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.C.; Tarn, W.Y. RBM4 down-regulates PTB and antagonizes its activity in muscle cell-specific alternative splicing. J. Cell. Biol. 2011, 193, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, M.P.; Nagel, R.J.; Fagg, W.S.; Shiue, L.; Cline, M.S.; Perriman, R.J.; Donohue, J.P.; Ares, M., Jr. Quaking and PTB control overlapping splicing regulatory networks during muscle cell differentiation. RNA 2013, 19, 627–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois, C.F.; Mortreux, F.; Auboeuf, D. The multiple functions of RNA helicases as drivers and regulators of gene expression. Nat. Rev. Mol. Cell Biol. 2016, 17, 426–438. [Google Scholar] [CrossRef]
- Dvinge, H. Regulation of alternative mRNA splicing: Old players and new perspectives. FEBS Lett. 2018, 592, 2987–3006. [Google Scholar] [CrossRef] [PubMed]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [Green Version]
- Appocher, C.; Mohagheghi, F.; Cappelli, S.; Stuani, C.; Romano, M.; Feiguin, F.; Buratti, E. Major hnRNP proteins act as general TDP-43 functional modifiers both in Drosophila and human neuronal cells. Nucleic Acids Res. 2017, 45, 8026–8045. [Google Scholar] [CrossRef] [Green Version]
- Militello, G.; Hosen, M.R.; Ponomareva, Y.; Gellert, P.; Weirick, T.; John, D.; Hindi, S.M.; Mamchaoui, K.; Mouly, V.; Döring, C.; et al. A novel long non-coding RNA Myolinc regulates myogenesis through TDP-43 and Filip1. J. Mol. Cell. Biol. 2018, 10, 102–117. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [Green Version]
- Nakamori, M.; Kimura, T.; Fujimura, H.; Takahashi, M.P.; Sakoda, S. Altered mRNA splicing of dystrophin in type 1 myotonic dystrophy. Muscle Nerve 2007, 36, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Pervouchine, D.D.; Knowles, D.G.; Guigó, R. Intron-centric estimation of alternative splicing from RNA-seq data. Bioinformatics 2013, 29, 273–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.G.; Bentley, D.R.; Bobrow, M. Infidelity in the structure of ectopic transcripts: A novel exon in lymphocyte dystrophin transcripts. Hum. Mutat. 1993, 2, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Reiss, J.; Rininsland, F. An explanation for the constitutive exon 9 cassette splicing of the DMD Gene. Hum. Mol. Genet. 1994, 3, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Gazzoli, I.; Pulyakhina, I.; Verwey, N.E.; Ariyurek, Y.; Laros, J.F.; ‘t Hoen, P.A.; Aartsma-Rus, A. Non-sequential and multi-step splicing of the dystrophin transcript. RNA Biol. 2016, 13, 290–305. [Google Scholar] [CrossRef] [Green Version]
- Castle, J.C.; Zhang, C.; Shah, J.K.; Kulkarni, A.V.; Kalsotra, A.; Cooper, T.A.; Johnson, J.M. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008, 40, 1416–1425. [Google Scholar] [CrossRef] [Green Version]
- Llorian, M.; Smith, C.W. Decoding muscle alternative splicing. Curr. Opin. Genet. Dev. 2011, 21, 380–387. [Google Scholar]
- Tinsley, J.M.; Blake, D.J.; Davies, K.E. Apo-dystrophin-3: A 2.2 kb transcript from the DMD locus encoding the dystrophin glycoprotein binding site. Hum. Mol. Genet. 1993, 2, 521–524. [Google Scholar] [CrossRef]
- Sibley, C.; Blazquez, L.; Ule, J. Lessons from non-canonical splicing. Nat. Rev. Genet. 2016, 17, 407–421. [Google Scholar] [CrossRef]
- Le Rumeur, E.; Winder, S.J.; Hubert, J.F. Dystrophin: More than just the sum of its parts. Biochim. Biophys. Acta 2010, 1804, 1713–1722. [Google Scholar] [CrossRef]
- Bies, R.D.; Phelps, S.F.; Cortez, M.D.; Roberts, R.; Caskey, C.T.; Chamberlain, J.S. Human and murine dystrophin mRNA transcripts are differentially expressed during skeletal muscle, heart, and brain development. Nucleic Acids Res. 1992, 20, 1725–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Górecki, D.C.; Monaco, A.P.; Derry, J.M.; Walker, A.P.; Barnard, E.A.; Barnard, P.J. Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum. Mol. Genet. 1992, 1, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.C.; Morris, G.E.; Howard, P.L.; Klamut, H.J.; Ray, P.N. Expression and synthesis of alternatively spliced variants of Dp71 in adult human brain. Neuromuscul. Disord. 2000, 10, 187–193. [Google Scholar] [CrossRef]
- Aragón, J.; Martínez-Herrera, A.; Romo-Yáñez, J.; Ceja, V.; Azotla-Vilchis, C.; Siqueiros-Márquez, L.; Soid-Raggi, G.; Herrera-Salazar, A.; Montañez, C. Identification of Dp71 Isoforms Expressed in PC12 Cells: Subcellular Localization and Colocalization with β-Dystroglycan and α1-Syntrophin. J. Mol. Sci. 2016, 58, 201–209. [Google Scholar] [CrossRef]
- Naidoo, M.; Anthony, K. Dystrophin Dp71 and the Neuropathophysiology of Duchenne Muscular Dystrophy. Mol. Neurobiol. 2020, 57, 1748–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greener, M.J.; Sewry, C.A.; Muntoni, F.; Roberts, R.G. The 3′-untranslated region of the dystrophin gene-conservation and consequences of loss. Eur. J. Hum. Genet. 2020, 10, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traverso, M.; Assereto, S.; Baratto, S.; Iacomino, M.; Pedemonte, M.; Diana, M.C.; Ferretti, M.; Broda, P.; Minetti, C.; Gazzerro, E.; et al. Clinical and molecular consequences of exon 78 deletion in DMD gene. J. Hum. Genet. 2018, 63, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.P.; Pletnikova, O.; Troncoso, J.C.; Wong, P.C. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 2015, 349, 650–655. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, J.; Emmett, W.; Fratta, P.; Isaacs, A.M.; Plagnol, V. Quantitative analysis of cryptic splicing associated with TDP-43 depletion. Bmc Med. Genom. 2017, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Quesnel-Vallières, M.; Weatheritt, R.J.; Cordes, S.P.; Blencowe, B.J. Autism spectrum disorder: Insights into convergent mechanisms from transcriptomics. Nat. Rev. Genet. 2019, 20, 51–63. [Google Scholar] [CrossRef]
- Li, Y.I.; Sanchez-Pulido, L.; Haerty, W.; Ponting, C.P. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 2015, 25, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ustianenko, D.; Weyn-Vanhentenryck, S.M.; Zhang, C. Microexons: Discovery, regulation, and function. Wires Rna 2017, e1418. [Google Scholar] [CrossRef] [PubMed]
- Doorenweerd, N.; Mahfouz, A.; van Putten, M.; Kaliyaperumal, R.; T’ Hoen, P.A.C.; Hendriksen, J.G.M.; Aartsma-Rus, A.M.; Verschuuren, J.J.G.M.; Niks, E.H.; Reinders, M.J.T.; et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci. Rep. 2017, 7, 12575. [Google Scholar] [CrossRef] [PubMed]
- Daoud, F.; Angeard, N.; Demerre, B.; Martie, I.; Benyaou, R.; Leturcq, F.; Cossée, M.; Deburgrave, N.; Saillour, Y.; Tuffery, S.; et al. Analysis of Dp71 contribution in the severity of mental retardation through comparison of Duchenne and Becker patients differing by mutation consequences on Dp71 expression. Hum. Mol. Genet. 2009, 18, 3779–3794. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.J.; Betts, G.A.; Maroulis, S.; Gilissen, C.; Pedersen, R.L.; Mowat, D.R.; Johnston, H.M.; Buckley, M.F. Dystrophin gene mutation location and the risk of cognitive impairment in Duchenne muscular dystrophy. PLoS ONE 2010, 5, e8803. [Google Scholar] [CrossRef]
- Mamchaoui, K.; Trollet, C.; Bigot, A.; Negroni, E.; Chaouch, S.; Wolff, A.; Kandalla, P.K.; Marie, S.; Di Santo, J.; St Guily, J.L.; et al. Immortalized pathological human myoblasts: Towards a universal tool for the study of neuromuscular disorders. Skelet. Muscle 2011, 1, 34. [Google Scholar] [CrossRef] [Green Version]
- Miro, J.; Laaref, A.M.; Rofidal, V.; Lagrafeuille, R.; Hem, S.; Thorel, D.; Méchin, D.; Mamchaoui, K.; Mouly, V.; Claustres, M.; et al. FUBP1: A new protagonist in splicing regulation of the DMD gene. Nucleic Acids Res. 2015, 43, 2378–2389. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Holt, I.; Mittal, S.; Furling, D.; Butler-Browne, G.S.; Brook, J.D.; Morris, G.E. Defective mRNA in myotonic dystrophy accumulates at the periphery of nuclear splicing speckles. Genes Cells 2007, 12, 1035–1048. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miro, J.; Bougé, A.-L.; Murauer, E.; Beyne, E.; Da Cunha, D.; Claustres, M.; Koenig, M.; Tuffery-Giraud, S. First Identification of RNA-Binding Proteins That Regulate Alternative Exons in the Dystrophin Gene. Int. J. Mol. Sci. 2020, 21, 7803. https://doi.org/10.3390/ijms21207803
Miro J, Bougé A-L, Murauer E, Beyne E, Da Cunha D, Claustres M, Koenig M, Tuffery-Giraud S. First Identification of RNA-Binding Proteins That Regulate Alternative Exons in the Dystrophin Gene. International Journal of Molecular Sciences. 2020; 21(20):7803. https://doi.org/10.3390/ijms21207803
Chicago/Turabian StyleMiro, Julie, Anne-Laure Bougé, Eva Murauer, Emmanuelle Beyne, Dylan Da Cunha, Mireille Claustres, Michel Koenig, and Sylvie Tuffery-Giraud. 2020. "First Identification of RNA-Binding Proteins That Regulate Alternative Exons in the Dystrophin Gene" International Journal of Molecular Sciences 21, no. 20: 7803. https://doi.org/10.3390/ijms21207803