





Abstract
Mammalian androgenones have two paternally or sperm-derived genomes. In mice (Mus musculus) they die at peri-implantation due to the misexpression of imprinted genes—the genes that are expressed monoallelically according to the parent of origin. The misexpressions involved are poorly defined. To gain further insight, we examined the causes of midgestation death of embryos with paternal duplication (PatDp) of distal chromosome 7 (dist7), a region replete with imprinted genes. PatDp(dist7) embryos have a similar phenotype to mice with a knockout of a maternally expressed imprinted gene, Ascl2 [achaete-scute complex homolog-like 2 (Drosophila)], and their death at midgestation could result from two inactive paternal copies of this gene. However, other dist7 misexpressions could duplicate this phenotype, and the potential epistatic load is undefined. We show that an Ascl2 transgene is able to promote the development of PatDp(dist7) embryos to term, providing strong evidence that Ascl2 is the only imprinted gene in the genome for which PatDp results in early embryonic death. While some of the defects in perinatal transgenic PatDp(dist7) fetuses were consistent with known misexpressions of dist7 imprinted genes, the overall phenotype indicates a role for additional undefined misexpressions of imprinted genes. This study provides implications for the human imprinting-related fetal overgrowth disorder, Beckwith–Wiedemann syndrome.
IN mice, Mus musculus, partheno- and androgenones, with two maternal and paternal genomes, respectively, die early at the peri-implantation stage. This developmental failure involves genomic imprinting, an epigenetic system in mammals resulting in the parent-specific monoallelic expression of a small subset of genes, many of which are crucial for normal growth and development. Parthenogenones overexpress maternally, and lack expression of paternally expressed imprinted genes, while the opposite misexpressions occur in androgenones (Bartonet al. 1984; Mann and Lovell-Badge 1984; McGrath and Solter 1984; Suraniet al. 1984; Cattanach and Kirk 1985; Coanet al. 2005; Fowdenet al. 2006). Understanding how the various misexpressions combine to bring about the death of partheno- and androgenones is important for understanding the etiology of genomic imprinting (Solter 2006; Wood and Oakey 2006; Renfreeet al. 2009) and the prevalence of sexual reproduction (Avise 2008).
An important region in terms of genomic imprinting is distal chromosome 7 (dist7), which contains >20 imprinted genes and at least two imprinting control regions (ICRs) (Schulzet al. 2006; Williamsonet al. 2010) (Figure 1). The Igf2/H19 (insulin-like growth factor 2/H19 fetal liver mRNA) ICR, or ICR1, functions as a DNA methylation-sensitive CTCF (CCCTC-binding factor)-based chromatin insulator. On the maternal chromosome, ICR1 binds CTCF and blocks Igf2 promoter–enhancer interaction. On the paternal chromosome, ICR1 is hyper-methylated, which prevents CTCF binding. Igf2 promoter–enhancer interaction is therefore allowed (Bell and Felsenfeld 2000; Harket al. 2000; Szabóet al. 2000). Telomeric to the ICR1 domain is a large cluster of imprinted genes under regulatory control of the KvDMR1 ICR, or ICR2. The active state of maternally derived genes within this cluster coincides with maternal-specific ICR2 hyper-methylation and the inactive state of the Kcnq1ot1 (KCNQ1 overlapping transcript 1) gene promoter contained within ICR2. The paternal ICR2 is hypo-methylated, and paternal-specific elongation of the Kcnq1ot1 transcript is associated with silencing of genes within the cluster in cis (Fitzpatricket al. 2002; Mancini-Dinardoet al. 2006; Shinet al. 2008).
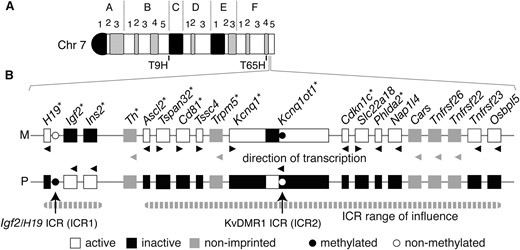
Imprinted genes regulated by ICR1 and ICR2. (A) Ideogram of G-banded mouse chromosome 7. The T9H and T65H breakpoints are at the B5/C border and F4, respectively, while the imprinted genes regulated by ICR1 and ICR2 are at F4/5. (B) All known genes within the two ICR clusters, imprinted and non-imprinted, are shown. Maternal (M) and paternal (P) chromosome. Genes for which knockouts have been published are indicated with an asterisk. Adapted partly from Beecheyet al. (1997) and Hudsonet al. (2010).
Research with mice with maternal and paternal duplication (MatDp and PatDp) of chromosome regions has identified regions of the genome subject to imprinting (Williamsonet al. 2010). Mice with MatDp and PatDp of dist7, as defined by the T(7;15)9H (T9H) and T(7;11)65H (T65H) reciprocal translocation breakpoints, die perinatally and early in gestation, respectively (Searle and Beechey 1990; McLaughlinet al. 1996; Beecheyet al. 1997). Mice with MatDp can be rescued through introduction of a mutant ICR1, which corrects misexpressions associated with two maternally derived ICRs, with Igf2 activation probably crucial (Hanet al. 2010). The cause of death of PatDp(dist7) embryos is unknown. The phenotype of PatDp(dist7)T9H embryos appears identical to mice with a null mutation of the dist7 imprinted gene Ascl2 [achaete-scute complex homolog-like 2 (Drosophila)], encoding a transcription factor essential for proliferation of placental spongiotrophoblast (Guillemotet al. 1995; McLaughlinet al. 1996). Maternal-specific expression of this and many other genes, including Cdkn1c [cyclin-dependent kinase inhibitor 1C (P57)], is regulated by ICR2 (Fitzpatricket al. 2002). While PatDp(dist7)T9H death is consistent with the presence of two paternally derived and epigenetically inactive copies of Ascl2 (McLaughlinet al. 1996), other misexpressions could duplicate this phenotype. Furthermore, the epistatic load of other dist7 misexpressions is likely to be considerable. Indeed, studies of PatDp(dist7)T9H embryonic stem (ES) cell chimeras revealed a potent negative effect of these cells on fetal development, akin to chimeras produced with androgenetic ES cells. Chimeras died at the fetal stage and possessed marked overgrowth of liver and heart and abdominal and skeletal defects (McLaughlinet al. 1997). Dist7 is likely to be an important influence in terms of failed androgenetic development as extensive and essentially complete genome-wide analysis has revealed that this is the only PatDp resulting in death at an early embryonic stage (Cattanach and Beechey 1997; Williamsonet al. 2010).
To better define how imprinted genes at the localized level of dist7 work together in regulating normal development, and to provide further insight into the developmental failure of androgenones, we examined some of the misexpressions of imprinted genes that might cause the developmental failure of PatDp(dist7)T9H conceptuses. We tested the developmental potential of PatDp(dist7)T9H embryos for which expression of Ascl2 was restored through the introduction of an Ascl2 transgene. This transgene resulted in the survival of PatDp(dist7)T9H embryos to late gestation with occasional live births. Nevertheless, defects were still observed with other misexpressions coming into play. Overexpression of Igf2 was corrected through the introduction of an Igf2 null mutation, which had some ameliorating effect. These findings provide implications for the human congenital imprinting-related disorder, Beckwith–Wiedemann syndrome, which is often associated with PatDp of the orthologous chromosome region.
MATERIALS AND METHODS
Mouse lines:
The following indicates the line, genotype, strain, source, and how it was produced and maintained: line 1—Tyr+/+, 129S1/SvImJ, The Jackson Laboratory (stock no. 002448); line 2—Tyrc/c, outbred Swiss CF-1, Charles River Laboratories; line 3—T9H/T9H, Tyr+/+, mix of C57BL/6J and CBA/Ca, The Jackson Laboratory (stock no. 001752); line 4—T9H/T9H, Tyrc/c, mix of C57BL/6J, CBA/Ca, and CF-1 (made by mating lines 2 with 3 and then intercrossing); line 5—Ascl2+/−, a mix of 129/Sv, outbred Swiss CD-1 and CF-1 [made by mating Ascl2+/− males (Guillemotet al. 1995) with females of line 2]. The mutation was maintained in (Ascl2+/+ ♀ × Ascl2+/− ♂) matings; line 6—0/Tg, Tyrc/c, a mix of C57BL/6J, CBA/Ca and CF-1 (made by producing founder transgenic lines as described below, then by crossing and backcrossing to line 2). The transgenes were maintained in (0/0 ♀ × 0/Tg ♂) matings; line 7—Igf2−/−, Tyrc/c, a mix of 129/SvEv and CF-1 [made by mating Igf2+/− mice (DeChiaraet al. 1990) with line 2, then by intercrossing]; line 8—Cdkn1c+/−, a mix of 129S7/SvEvBrd (129S7), C57BL/6J and CF-1 [made by mating Cdkn1c+/− males (Zhanget al. 1997) with females of line 2]. The mutation was maintained in (Cdkn1c+/+ ♀ × Cdkn1c+/− ♂) matings; line 9—FVB/NJ.CAST/Ei(N3), which is homozygous for the M. musculus castaneus form of dist7 (Szabóet al. 2002).
Production of Ascl2 transgenic mice:
A genomic DNA clone containing the Ascl2 gene was obtained by screening a P1 genomic DNA library—mouse strain 129XI/SvJ, vector pAD10SacBII (Genome Systems). Ends were located at 17.64 kb 5′ and 66.34 kb 3′ of the Ascl2 ATG translation initiation codon. One end of the 84-kb insert was at 19 kb from the nearest annotated gene toward the telomere, Tspan32 (tetraspanin 32). The other end was within the first intron of the nearest annotated gene toward the centromere, Th (tyrosine hydroxylase). Founder transgenic mice were made using the whole Ascl2 P1 clone in circular form (C) or after linearization (L) with NotI [e.g., founders Tg(Ascl2)6C and -10L, respectively]. Zygotes of strain (C57BL/6J × CBA/CaJ)F2 were microinjected with DNA as described (Mann and McMahon 1993; Mann 1993) to obtain founder animals identified by PCR using primers specific for the pAD10SacBII vector sequence, 5′-TCTG GCAG GAGG AGCG ACTC AAG-3′ and 5′-CCGT TTTC TGTT CCCG TG-3′ (753-bp amplicon). Copy number and chromosomal location of the integration events were not investigated. Transgenic integration events (Tg) were tested for function by introducing them into Ascl2 mutants. Paternal transmission was as follows: (Ascl2+/−, 0/0 ♀ × Ascl2+/+, 0/Tg ♂) matings, testing for survival to term of (Ascl2−/+, 0/Tg) embryos. Maternal transmission was as follows: (Ascl2+/−, Tg/0 ♀ × Ascl2+/+, 0/0 ♂) matings, testing for survival to term of (Ascl2−/+, Tg/0) embryos.
Matings:
Production of PatDp(dist7)T9H-0/Tg, Igf2+/+ fetuses:
Female (T9H/+, Tyr+/+, 0/0, Igf2+/+) parents were bred in (line 3 ♀ × line 1 ♂ and reciprocal matings). Male (T9H/+, Tyrc/c, Tg/0, Igf2+/+) parents were bred in (line 6 ♀ × line 4 ♂) matings. Fetuses were a mix of strains C57BL/6J, CBA/Ca, and CF-1.
Production of PatDp(dist7)T9H-0/Tg, Igf2+/− and recombinant -Igf2−/− and -Igf2+/+ fetuses:
Female (T9H/+, Tyr+/+, 0/0) parents were bred in (line 3 ♀ × line 1 ♂ and reciprocal matings). Male (T9H/+, Tyrc/c, Tg/0, Igf2−/+) parents were bred in [(line 7 ♀ × line 6 ♂) ♀ × line 4 ♂] matings. Fetuses were a mix of strains 129S1/SvImJ, C57BL/6J, CBA/Ca, 129/SvEv, and CF-1.
Genotyping:
The three Igf2 genotypes were identified by using two PCR assays, the first detecting the mutant allele, 5′-AGCC ATTC TCCT GGGA TTAG GG-3′ (intronic) and 5′-AGCA GCCG ATTG TCTG TTGT GC-3′ [within neo coding sequence (cds)] (∼450-bp amplicon), and the second detecting the wild-type allele, 5′-TACA GTTC AAAG CCAC CACG G-3′ (intronic) and 5′-GCCA AAGA GATG AGAA GCAC CAAC-3′ (exonic) (323-bp amplicon). The Ascl2− allele was identified using primers, 5′-GCCT TCTA TCGC CTTC TTGA CG-3′ (within neo cds) and 5′-CCCC CTAA CCAA CTGG AAAA GTC-3′ (exonic) (∼650-bp amplicon). The Cdkn1c− allele was identified using primers specific for the selection cassette used in gene targeting, 5′-CTCA GAGG CTGG GAAG GGGT GGGT C-3′ [within Pgk1 (phosphoglycerate kinase 1) promoter] and 5′-ATAC TTTC TCGG CAGG AGCA AGGT G-3′ (within neo cds) (520-bp amplicon). The Ascl2 transgene was identified as described above.
Gene and protein expression:
Northern blots were performed as described (Szabó and Mann 1994; McLaughlinet al. 1996). Probes were made using the following primers: Ascl2, made by RT–PCR, 5′-GGCT GTTA ACAC CCGC TACT C-3′ and 5′-AGCA CTTG GCAT TTGG TCAG-3′ (356-bp amplicon); Cdkn1c, made by RT–PCR, 5′-GCCG GGTG ATGA GCTG GGAA-3′, and 5′-AGAG AGGC TGGT CCTT CAGC-3′ (221-bp amplicon); Igf2 major transcript, made by RT–PCR, 5′-CCTC TCCC GTCC CTCA GTGT CA-3′ and 5′-TGTC CAGC CAAA TGGG CAGG TA-3′ (558-bp amplicon). Probes for H19 and Gapdh (glyceraldehyde-3-phosphate dehydrogenase) transcripts were as described (Szabó and Mann 1994). Probes were hybridized independently to the same blots after stripping. Radioactivity of bands was quantified using a Typhoon biomolecular imager (GE Healthcare). For Ascl2 quantification, values were normalized to the Gapdh value.
Quantitative RT–PCR single-nucleotide primer extension (SNuPE) assay for Ascl2, which relies on the presence of a single-nucleotide polymorphism present in an exon, was performed as described (Szabó and Mann 1995). Primers used in RT–PCR were exonic and spanned a single intron: 5′-CATA TTTT CAGT AGAG TCCT ACA-3′ and 5′-GGGA CAGA GGTC ATCT TTAT TGTG C-3′. PCR conditions were the following: (95°/45 sec, 56°/45 sec, 72°/1 min) × 33 cycles (351-bp amplicon). The SNuPE primer was 5′-CATA TTTT CAGT AGAG TCCT ACA-3′ and incorporated a radionucleotide during SNuPE, strain 129XI/SvJ—A × 1 and strain CAST/Ei—G × 1. Radioactivity of bands was quantified using a biomolecular imager as described above.
IGF2 protein concentration was measured by radioimmunoassay after removal of IGF-binding proteins by BioSpin separation, using Bio-Gel P10 (Bio-Rad) in the presence of 1 m acetic acid (Mohan and Baylink 1995). This method has been validated for measurement of IGF2 in mouse serum (Miyakoshiet al. 2001). Skeletons were prepared and stained with Alizarin Red S (bone) and Alcian Blue 8GX (cartilage) as described (McLeod 1980). Histological sections were stained with hematoxylin and eosin.
RESULTS
Production of a functional Ascl2 transgene:
A validated non-imprinted Ascl2 transgene was required for restoration of Ascl2 activity in PatDp(dist7)T9H embryos. Approximately 120 zygotes were injected with the Ascl2 P1 clone from which six transgenic lines were derived. In all cases, the transgene (Tg) was transmitted in Mendelian ratios. Each independent integration event was tested for function using matings as described in materialsandmethods. For paternal transmission, three integration events rescued mutants at the frequency expected for survival of every (Ascl2−/+, 0/Tg) individual, or one-third of the neonates genotyped: Tg(Ascl2)2C, -6C, and -10L; 12/33 (36%), 11/23 (48%), and 15/44 (34%) rescued, respectively. For the remaining three lines, very low or zero frequency of survival was observed. In these cases, it is probable that the transgene was expressed at a low level relative to the endogenous gene. For maternal transmission, for each of the six lines, the frequency of survival of (Ascl2−/+, Tg/0) embryos to term was equivalent to that obtained on paternal transmission. For the three rescuing lines, these latter results show that the transgene segregated independently of Ascl2. Overall, the results indicate that none of the integration events were subject to imprinting, this being expected given that imprinting of Ascl2 is regulated by ICR2 and not by immediately adjacent sequences.
Expression of Ascl2 was examined in two of the three rescuing lines—Tg(Ascl2)6C and -10L. In 15½ days post-coitum (dpc) transgenic placentas, Northern blots showed that the total amount of Ascl2 RNA was 7.7 (line -6C) and 4.3 (line -10L) times greater than endogenous Ascl2 RNA (Figure 2A). In 10½ dpc Tg(Ascl2)10L transgenic placentas, quantitative RT–PCR SNuPE assays showed that ∼75% of the total placental Ascl2 RNA was derived from the transgene, regardless of parental inheritance (Figure 2B). Together, these results indicate that in line Tg(Ascl2)10L, from 10½–15½ dpc, total Ascl2 RNA is approximately fourfold higher than endogenous, with three-fourths of this RNA derived from the transgene. The Tg(Ascl2)10L transgene was selected for introduction into PatDp(dist7)T9H embryos.
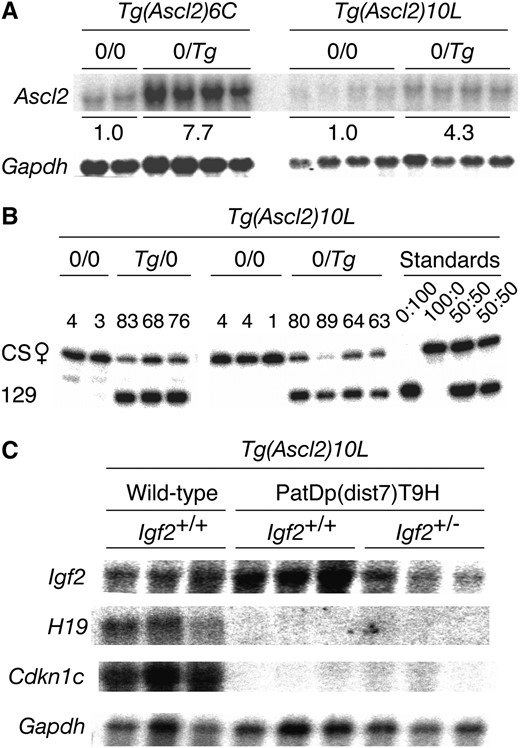
Ascl2 expression in placentas. (A) Northern blot. Placentas at 15½ dpc from (Ascl2+/+, 0/0 ♀ × Ascl2+/+, 0/Tg ♂) matings. Values under the bands represent the average relative amount of total Ascl2 RNA corrected for the amount of Gapdh RNA determined after washing and rehybridization. (B) RT–PCR SNuPE assay to determine the ratio of allele-specific expression. Placentas at 10½ dpc from (Ascl2+/+, 0/0 ♀ × Ascl2+/+, 0/Tg ♂) and reciprocal matings. The endogenous maternal and paternal Ascl2 alleles were derived from strain CAST/Ei (CS) and 129, respectively, while the Tg was also derived from strain 129. Values above each pair of bands represent the amount of 129-derived Ascl2 RNA as the percentage of the total amount of Ascl2 RNA present. In Tg/0 (Tg maternally derived) and 0/Tg (Tg paternally derived) embryos, the amount of Tg-derived RNA is on average 76% and 74% of the total, respectively. CS:129 placenta RNA. FVB/NJ.CS(N3) females homozygous for the CS form of dist7 (Szabóet al. 2002) were used in matings described above and as a source of RNA. (C) Northern blot. Placentas at 17½–18½ dpc. Three of each genotype were used, as shown.
Rescue of PatDp(dist7)T9H embryos by the Ascl2 transgene and expression of imprinted genes:
To produce PatDp(dist7)T9H zygotes, (T9H/+, Tyr+/+, 0/0 ♀ × T9H/+, Tyrc/c, Tg/0 ♂) matings were performed (Figure 3A). One in two PatDp(dist7)T9H zygotes would inherit Tg and not be subject to ASCL2 deficiency. If viable, they would be identifiable from 12½ days dpc as albinos, lacking in eye and fur pigmentation, as they must be homozygous for the dist7 marker, albino (c), a mutation of the Tyr (tyrosinase) gene. At 13½–18½ dpc, albino or PatDp(dist7)T9H fetuses were observed at approximately the frequency expected—one in 15—and all carried the Ascl2 transgene. These PatDp(dist7)T9H-0/Tg, Tyrc/c fetuses would be expected to have two active alleles of Igf2 and to overexpress this gene. To normalize the level of Igf2 transcript, (T9H/+, Tyr+/+, 0/0, Igf2+/+ ♀ × T9H/+, Tyrc/c, Tg/0, Igf2−/+ ♂) matings were also performed, which would produce PatDp(dist7)T9H-0/Tg, Tyrc/c, Igf2+/− and also recombinant -Igf2+/+ and -Igf2−/− zygotes (Figure 3B).
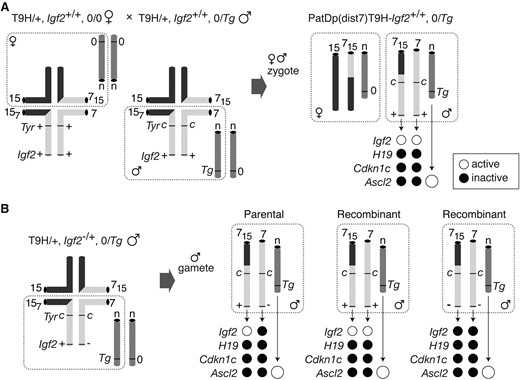
Production of PatDp(dist7)T9H fetuses and allele-specific expression. (A) Quadrivalents at meiosis in translocation heterozygotes (T9H/+). Male parent is Igf2+/+, Tyrc/c, or albino and Tg/0 with Tg on an unknown autosome (n). The female parent is Tyr+/+ and does not carry Tg (0/0). Unbalanced complementary gametes (dashed boundaries) combine to give PatDp(dist7)T9H-0/Tg zygotes identified as albinos. Half of the PatDp(dist7)T9H zygotes will be 0/0 with respect to Tg and therefore cannot develop beyond 10½ dpc. Expected activity state of an allele is indicated by a solid circle (inactive) or an open circle (active). Expression of Ascl2 derived from Tg is greater than for the endogenous allele (larger circles). (B) Quadrivalent at meiosis in male T9H/+, Igf2+/− parents, which were mated to the same females as in A. The three types of gametes produced are depicted: Igf2+/− (parental) and Igf2+/+ or Igf2−/− (recombinants). Chromosomes depicted are actually paired chromatids. Other details as in A.
In late-term PatDp(dist7)T9H-0/Tg fetuses that were recovered, the expression levels of Igf2, H19, and Cdkn1c in the placenta were assessed in Northern blots. As expected, PatDp(dist7)T9H-0/Tg, Igf2+/+, and -Igf2+/− placentas lacked H19 and Cdkn1c expression, with the former apparently having excess Igf2 expression, and the latter, a normal amount of transcript (Figure 2C). Fetal serum IGF2 protein concentrations reflected the level of transcript and are expressed in nanograms per microliter: controls—four wild-type Igf2+/+ fetuses (153, 153, 147, and 174), three mutant Igf2+/− fetuses lacking Igf2 RNA (60, 63, 66), equivalent to background concentration; experimentals—two PatDp(dist7)T9H, 0/Tg, Igf2+/+ fetuses (180, 186), two PatDp(dist7)T9H-0/Tg, Igf2+/− fetuses (120, 138), and one PatDp(dist7)T9H-0/Tg, Igf2−/− fetus (48). Therefore PatDp(dist7)T9H, 0/Tg, Igf2+/+ and -Igf2+/− fetuses had ∼1.3- and 0.7-fold the normal concentration of serum IGF2, respectively. Despite the high frequency of rescue of PatDp(dist7)T9H fetuses at late term by the Ascl2 transgene, the phenotype of these fetuses was not completely normalized as the effects of other misexpressions had come into play.
Defects in perinatal PatDp(dist7)T9H, 0/Tg fetuses and newborns:
The expected frequency of recovery of PatDp(dist7)T9H-0/Tg zygotes is ∼1 in 50 and is further complicated by small litter sizes due to the death of the majority of ova at peri-implantation due to chromosome imbalance. It is therefore difficult to obtain sufficient numbers and material for in-depth analysis of phenotype. Nevertheless, consistent abnormalities were observed. While wild-type placenta carrying Tg were normal in appearance (Figure 4A), PatDp(dist7)T9H-0/Tg, Igf2+/+ displayed a pronounced placentamegaly, often being at least twofold heavier with increased proportions compared to wild-type uterus mates (Figure 5, E and F, Figure 6). Placentamegaly was also seen in PatDp(dist7)T9H-0/Tg, Igf2+/− and -Igf2−/− fetuses, although possibly was not as marked (Figure 6). Additional -Igf2+/− and Igf2−/− individuals were obtained that confirmed the presence of this defect (data not shown), and histological examination revealed a disorganization of labyrinth and spongiotrophoblast possibly due to the presence of greatly enlarged maternal blood sinuses filled with red blood cells (Figure 4, B and C).
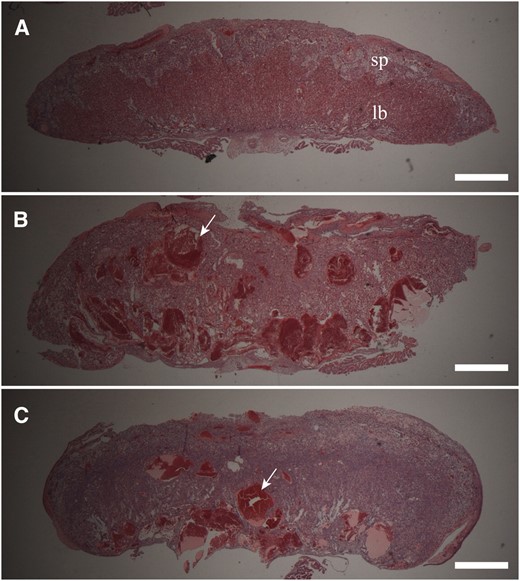
Placenta phenotype in PatDp(dist7)T9H-0/Tg fetuses. (A) Wild-type 0/Tg, Igf2+/+. sp, 17½ dpc spongiotrophoblast layer; lb, labyrinth trophoblast layer. (B) PatDp(dist7)T9H-0/Tg, Igf2+/− 17½ dpc. A portion of this placenta was removed for RNA extraction. (C) PatDp(dist7)T9H-0/Tg, Igf2−/−, 18½ dpc. Bars, 1 mm.
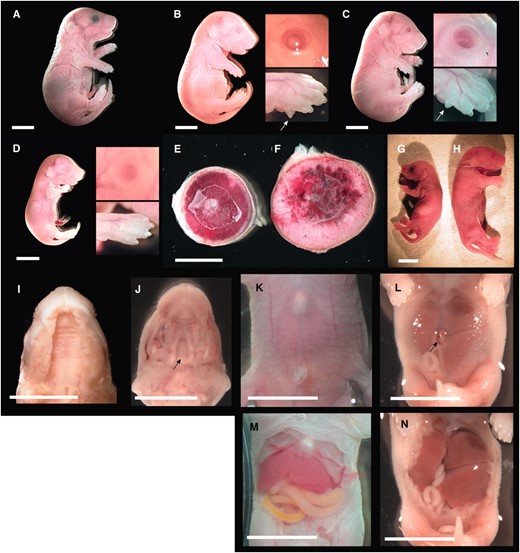
Overt phenotype of late-term PatDp(dist7)T9H-0/Tg fetuses. (A) Fetus, 18½ dpc, wild type. (B) Fetus, 18½ dpc, PatDp(dist7)T9H-0/Tg, Igf2+.+; insets show eye and forepaw, with arrow indicating additional digit or polydactyly. (C) Fetus, 18½ dpc, PatDp(dist7)T9H-0/Tg, Igf2+/−, insets and arrow as for B. (D) Fetus, 18½ dpc, PatDp(dist7)T9H-0/Tg, Igf2−/−, insets as for B. (E) Placenta, 16½ dpc, wild type. (F) Placenta, 16½ dpc, PatDp(dist7)T9H-0/Tg, Igf2+/+, uterus mate to E. (G) Neonate, ½ days post partum (dpp), Igf2+/−. (H) Neonate, ½ dpp, PatDp(dist7)T9H-0/Tg, Igf2+/−. (I) Exposed palate, 18½ dpc, PatDp(dist7)T9H-0/Tg, Igf2+/+. (J) Exposed palate, 17½ dpc, Cdkn1c−/+. Arrow indicates cleft in palate. (K) Abdomen, 18½ dpc, wild type. (L) Abdomen, 18½ dpc, PatDp(dist7)T9H-0/Tg, Igf2+/+. Arrow indicates hernia adjacent to the umbilicus. (M) Exposed abdomen of K. (N) Exposed abdomen of L. Bars, 0.5 cm.
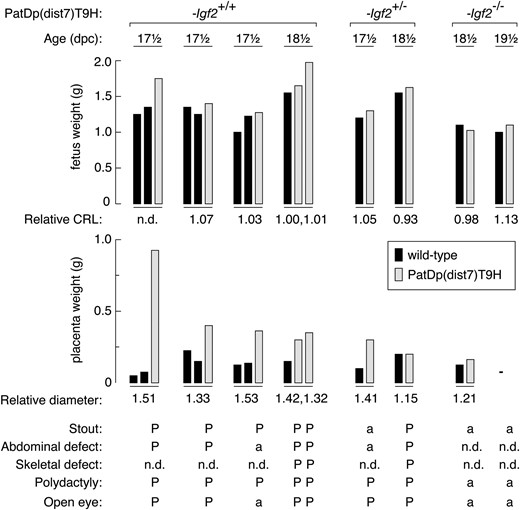
Summary of phenotypic defects in PatDp(dist7)T9H-0/Tg fetuses. PatDp(dist7)T9H fetuses (shaded bars), with Igf2 genotype as indicated at top, are grouped with wild-type uterus mates (solid bars). Phenotypic features for each individual are indicated from top to bottom. Relative CRL (crown-rump length) and relative diameter (for placenta) is the value obtained for PatDp(dist7)T9H divided by the value obtained for wild type. P, defect present; a, defect absent. n.d., not done; —, not applicable (the 19½ dpc PatDp(dist7)T9H-0/Tg, Igf2−/− individual and its wild-type littermate were newborns, and therefore the placenta was not measured).
All PatDp(dist7)T9H-0/Tg, Igf2+/+ and -Igf2+/− rescued fetuses were stout or thickset in appearance (Figure 5, B and C). Stoutness is consistent with CDKN1C deficiency, but the absence of this feature in all of the three PatDp(dist7)T9H-0/Tg, Igf2−/− fetuses recovered (Figure 5D) indicates that this defect is also dependent on the presence of IGF2. In PatDp(dist7)T9H-0/Tg, Igf2+/+ fetuses, abnormalities consistent with IGF2 excess included increased weight relative to wild-type uterus mates, although the small numbers obtained do not allow for statistical analysis of significance (Figure 6). They were not longer, as crown-rump length was not relatively increased (Figure 6). In PatDp(dist7)T9H-0/Tg, Igf2+/− fetuses, in which IGF2 concentrations were normalized, insufficient numbers were obtained to determine if they were of normal weight. Polydactyly was seen in every PatDp(dist7)T9H-0/Tg, Igf2+/+ and -Igf2+/− fetus recovered. Eyelid fusion failure or open eyes occurred in four of five PatDp(dist7)T9H-0/Tg, Igf2+/+ and in two of seven PatDp(dist7)T9H-0/Tg, Igf2+/− rescued fetuses. None of three PatDp(dist7)T9H-0/Tg, Igf2−/− fetuses possessed polydactyly or open eyes, indicating that these defects were dependent on IGF2. Some of these fetuses are further described (Figure 5, B–D, Figure 6).
Most PatDp(dist7)T9H-0/Tg, Igf2+/+ and -Igf2+/− fetuses possessed abdominal abnormalities in the form of thinned abdominal skin or muscle wall through which the liver and gut could be easily discerned. Often this was accompanied by a hernia adjacent to the umbilicus, with or without protrusion of intestines or omphalocele (Figure 5L). The distension of the abdomen in PatDp(dist7)T9H-0/Tg, Igf2+/+ fetuses appeared to be caused by an enlarged liver (Figure 5, L and N). In all viable PatDp(dist7)T9H-0/Tg, Igf2+/+ and -Igf2+/− fetuses and controls, the gut was removed and straightened. Unexpectedly, in all cases, it was of normal morphology and was not shortened as usually seen in Cdkn1c mutants (Yanet al. 1997; Zhanget al. 1997).
PatDp(dist7)T9H-0/Tg fetuses of all three Igf2 genotypes possessed shortened limbs (Figure 5, B and D); this defect was consistent with the reduced chondrogenesis associated with CDKN1C deficiency (Yanet al. 1997; Zhanget al. 1997). Unexpectedly, no PatDp(dist7)T9H-0/Tg fetus possessed cleft palate (Figure 5I), a common defect in Cdkn1c mutants (Figure 5J) (Yanet al. 1997; Zhanget al. 1997). Differential staining of bone and cartilage revealed a generalized hypo-ossification of the skull, vertebrae, limbs, and ribs accompanied by bifurcation of the sternum (Figure 7, E–H). Thus, reduced chondrogenesis and hypo-ossification are as penetrant in PatDp(dist7)T9H-0/Tg fetuses as in Cdkn1c mutants (Figure 7, I–L), although not as severe. The same severity of defects was seen in all of four additional PatDp(dist7)T9H-0/Tg fetuses and newborns—one -Igf2+/+ and three -Igf2+/− (data not shown).
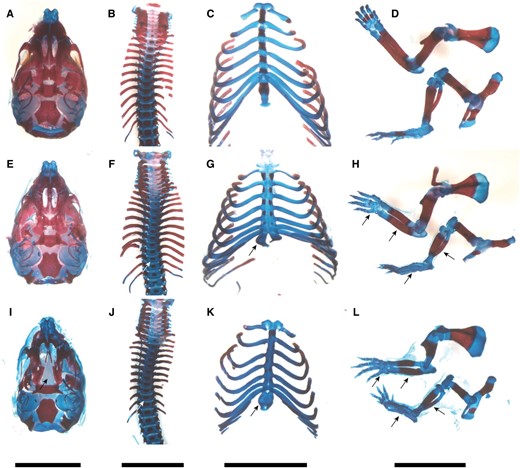
Phenotype of skeleton in late-term PatDp(dist7)T9H-0/Tg fetuses. (A–D) Wild type. (E–H) PatDp(dist7)T9H-Igf2+/−, 0/Tg. (I–L) Cdkn1c−/+. (A, E, and I) Skull, ventral surface. (B, F, and J) Spine. (C, G, and K) Ribcage. (D, H, and L) Forelimb (top); hindlimb (bottom). Bone, stained red; cartilage, stained blue. Prominent features, as indicated by arrows, include cleft palate (I), smaller ossification centers in vertebrae (F and J), bifurcated sternum (G and K), shorter bone length in forelimbs and hindlimbs and hypo-ossification in phalanges (H and L). Bars, 0.5 cm.
DISCUSSION
When provided with a functional Ascl2 transgene, PatDp(dist7)T9H embryos were able to develop to the latest stages of gestation, providing strong evidence that the death of PatDp(dist7)T9H embryos at midgestation is the result of two epigenetically inactivated endogenous copies of the Ascl2 gene. A contribution from other misexpressions in their death cannot be entirely ruled out, as the Ascl2 transgene used was overexpressed and in other ways may not have been expressed exactly in accord with the endogenous gene. Such nonphysiological Ascl2 expression could compensate in some way for other existent misexpressions. The degree of survival after restoration of Ascl2 expression is remarkable, given the potent effects of PatDp(dist7)T9H ES cells in chimeras (McLaughlinet al. 1997). The present experiments extend these previous observations of chimeras by allowing for the assessment of the effects of PatDp(dist7)T9H when non-mosaic, of the contribution of excess IGF2 to the phenotype, and of the effect of PatDp(dist7)T9H on placental development, which is not possible with ES cells as they do not colonize the trophoblast.
The phenotype of PatDp(dist7)T9H-0/Tg, Igf2+/+ fetuses results from the combined action of all misexpressed imprinted genes located at dist7 and is likely to have a complex etiology. For placentamegaly, a partial explanation is lack of Cdkn1c expression, as Cdkn1c mutant placentas are 1.4-fold heavier than wild type (Takahashiet al. 2000). In PatDp(dist7)T9H-0/Tg, Igf2+/+ fetuses, the greater increase in size, together with labyrinth or spongiotrophoblast disorganization and enlarged maternal blood sinuses, can be sufficiently explained by CDKN1C deficiency together with IGF2 excess: When this specific combination of misexpressions is brought about through genetic modification, a similar phenotype results (Casparyet al. 1999). Nevertheless, in PatDp(dist7)T9H fetuses, other misexpressions must be involved as placentamegaly persisted in PatDp(dist7)T9H-0/Tg, Igf2+/− and -Igf2−/− fetuses. A good candidate for this effect is the inactivity of the maternally expressed Phlda2 (pleckstrin homology-like domain, family A, member 2) gene, regulated by ICR2. This gene is a negative regulator of placental growth, with Phlda2 null mice having greatly enlarged placentas (Franket al. 2002). There are a number of other candidates, as most if not all imprinted genes under regulatory control of ICR2 are expressed in the placenta (Coanet al. 2005) and are likely to be inactive in PatDp(dist7)T9H-0/Tg conceptuses. Also, other dist7 imprinted genes are expressed in the placenta and are not regulated by ICR1 or ICR2 (Schulzet al. 2006). Finally, it is conceivable that the threefold extra Ascl2 RNA derived from the Ascl2 transgene may have contributed to the placental phenotype. However, wild-type fetuses carrying Tg had overtly normal placentas, as did Tg(Ascl2)6C fetuses in which expression of the transgene was at least sixfold greater than endogenous Ascl2 RNA.
That Tg(Ascl2)6C and -10L placentas were able to develop apparently normally in the presence of an appreciable excess of Ascl2 RNA suggests that precise regulation of ASCL2 is not crucial for trophoblast proliferation and differentiation. This finding is of interest given that previous studies have indicated that suppression of ASCL2 function is required for trophoblast giant cell (TGC) differentiation. While this suppression is thought to be brought about by the action of MDFI (MyoD family inhibitor) (Krautet al. 1998), experiments in cultured rat trophoblast cells indicate that excess ASCL2 can override the suppressive mechanism (Scottet al. 2000). Tg(Ascl2) mice could provide a useful tool to further investigate the importance of ASCL2 levels in TGC differentiation.
While occasional PatDp(dist7)T9H-0/Tg newborns were obtained, survival beyond this stage is very unlikely, given their phenotype. A number of defects seen in the PatDp(dist7)T9H-0/Tg fetus proper were consistent with CDKN1C deficiency, with abdominal defects, hypo-ossification, and short limbs prominent in PatDp(dist7)T9H-0/Tg, Igf2+/+, and -Igf2+/− fetuses. However, CDKN1C deficiency-associated defects of cleft palate and gut shortening were absent, regardless of Igf2 genotype. The reasons for these conspicuous deviations from the standard Cdkn1c null phenotype are unclear, but it is conceivable that a small number of cells located in important proliferation zones at least partially dispense with imprinting-regulated expression. If so, then Cdkn1c expression would be higher when two paternal wild-type alleles are present, PatDp(dist7)T9H, in comparison to one, Cdkn1c−/+. This is conceivable given that the imprinting is not always tightly regulated at the tissue-specific level, for example (Leeet al. 1997). That genetic background may have influenced the result is unlikely, given that cleft palate and gut abnormalities are penetrant in Cdkn1c mutants in a variety of mixed strain backgrounds (Yanet al. 1997; Zhanget al. 1997).
The defects of polydactyly and open eyes in PatDp(dist7)T9H-0/Tg, Igf2+/+, and -Igf2+/− fetuses are associated with excess IGF2. Polydactyly has been described in mice with biallelic expression of Igf2 (Eggenschwileret al. 1997; Szabóet al. 2004), and both defects are seen in fetuses with IGF2 super-excess; this occurs when biallelic expression of Igf2 is combined in the same fetus with lack of activity of the chromosome 17 Igf2r (insulin like growth factor 2 receptor) gene (Eggenschwileret al. 1997). Given the apparent dependency of these defects on excess IGF2, their occurrence was unexpected in PatDp(dist7)T9H-0/Tg, Igf2+/− fetuses in which IGF2 concentrations were normalized. Similar to placentamegaly, these results point to a role for misexpressions other than CDKN1C deficiency and IGF2 excess in the etiology, although these defects appear to have a partial requirement for IGF2 as they were not observed in PatDp(dist7)T9H-0/Tg, Igf2−/− fetuses. Hepato- and cardiomegaly were reported in PatDp(dist7)T9H ES cell chimeras (McLaughlinet al. 1997), and at least the former appeared to be present in PatDp(dist7)T9H-0/Tg, Igf2+/+ fetuses. Again, lack of expression of the negative growth regulator Phlda2 may play a role. Normally, Phlda2 is expressed and imprinted in liver, lung, and kidney, and while its lack of expression does not lead to a discernible phenotype in the fetus (Franket al. 2002), it might cause pathology when combined with other misexpressions.
This genetic analysis provides implications for Beckwith–Wiedemann syndrome, a disease commonly associated with sporadic epigenetic lesions at the dist7 orthologous chromosome region, 11p15.5. Defects observed are placentamegaly, macrosomia, organomegaly including macroglossia, abdominal defects, and a high incidence of childhood cancer. We note that ASCL2, while located at 11p15.5, is not imprinted in humans (Westermanet al. 2001) and therefore probably has no relevance for this disease. On the basis of mouse models (Eggenschwileret al. 1997; Sunet al. 1997; Casparyet al. 1999) and familial cases of the disease, it is thought that much of the syndrome can be explained by CDKN1C deficiency, IGF2 excess, or both (Lamet al. 1999; DeBaunet al. 2002; Cooperet al. 2005; Enklaaret al. 2006). As discussed above, much of the pathology in PatDp(dist7)T9H-0/Tg fetuses is consistent with the same etiology. However, the observed deviations from this theme are strongly indicative of roles for unknown misexpressions in the PatDp(dist7)T9H phenotype and imply that the same may be true for the human disease.
These findings show that Ascl2 is probably the only imprinted gene in the genome for which PatDp causes early embryonic death. This finding emphasizes and clarifies the concept that the peri-implantation death of androgenones is the synergistic result of a number of misexpressions, which remain undefined. There is evidence that disruption of the expression of one imprinted gene can affect the expression of others (Varraultet al. 2006; Gaboryet al. 2009). A parallel situation is likely to be true for parthenogenones. For MatDp, the earliest stage of death is brought about by MatDp of proximal chromosome 6 and is likely mediated by inactivity of the Peg10 (paternally expressed 10) gene. Like Ascl2 mutants and PatDp(dist7)T9H embryos, Peg10 mutants lack placental spongiotrophoblast (Onoet al. 2006; Williamsonet al. 2010). It is intriguing that, for MatDp and PatDp, the earliest acting single imprinted gene misexpression that results in a developmental barrier involves the silencing of a gene essential for the viability of the same tissue at the same developmental stage. In both cases, the extra-embryonic tissues could be primarily affected because partheno- and androgenetic epiblasts appear intrinsically viable, given that ES cell lines can be readily derived from either (Robertson 1987; Mannet al. 1990). It would seem advantageous, or at least convenient, for mice for imprinting to act to prevent the development of uniparental embryos beyond the peri-implantation stage, thereby conserving valuable reproductive resources.
Footnotes
Present address: Department of Neurosurgery, Cedars Sinai Medical Center, Los Angeles, CA 90048.
Present address: Department of Molecular and Cellular Biology, Beckman Research Institute, City of Hope National Medical Center, Duarte, CA 91010.
Footnotes
Communicating editor: T. R. Magnuson
Acknowledgements
We thank Andras Nagy for the Ascl2 knockout mouse line, Elizabeth Robertson and Agiris Efstratiadis for the Igf2 knockout mouse line, James Cross and Stephen Elledge for the Cdkn1c knockout mouse line, and Trevelyan Menheniott for stimulating discussions. This work was supported by the National Institutes of Health and the National Health and Medical Research Council of Australia.
References
Avise, J. C.,
Cattanach, B. M., and C. V. Beechey,
Enklaar, T., B. U. Zabel and D. Prawitt,
Fowden, A. L., C. Sibley, W. Reik and M. Constancia,
Frank, D., W. Fortino, L. Clark, R. Musalo, W. Wang et al.,
Lam, W. W., I. Hatada, S. Ohishi, T. Mukai, J. A. Joyce et al.,
Mohan, S., and D. J. Baylink,
Robertson, E. J.,
Williamson, C. M., A. Blake, S. Thomas, C. V. Beechey, J. Hancock et al.,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}