




Abstract
The nuclear-encoded Krebs cycle enzymes, fumarate hydratase (FH) and succinate dehydrogenase (SDHB, -C and -D), act as tumour suppressors. Germline mutations in FH predispose individuals to leiomyomas and renal cell cancer (HLRCC), whereas mutations in SDH cause paragangliomas and phaeochromocytomas (HPGL). In this study, we have shown that FH-deficient cells and tumours accumulate fumarate and, to a lesser extent, succinate. SDH-deficient tumours principally accumulate succinate. In situ analyses showed that these tumours also have over-expression of hypoxia-inducible factor 1α (HIF1α), activation of HIF1αtargets (such as vascular endothelial growth factor) and high microvessel density. We found no evidence of increased reactive oxygen species in our cells. Our data provide in vivo evidence to support the hypothesis that increased succinate and/or fumarate causes stabilization of HIF1α a plausible mechanism, inhibition of HIF prolyl hydroxylases, has previously been suggested by in vitro studies. The basic mechanism of tumorigenesis in HPGL and HLRCC is likely to be pseudo-hypoxic drive, just as it is in von Hippel–Lindau syndrome.
INTRODUCTION
Succinate dehydrogenase (SDH) and fumarate hydratase (FH) are housekeeping genes which encode the Krebs cycle proteins involved in the fundamental processes of energy production. These enzymes act in the cell's energy production system, and not surprisingly, their deficiency results in early onset, severe encephalopathy (1). However, the genes encoding both enzymes also act as tumour suppressors: germline mutations in the B, C and D subunits of SDH predispose individuals to hereditary paragangliomatosis with phaeochromocytomas (HPGL) (2–6) and germline mutations in FH cause hereditary leiomyomatosis and renal cell cancer (HLRCC) (7). Both of these familial cancer syndromes are inherited in an autosomal dominant manner, apparently with high penetrance. In HPGL, SDHD mutations are associated with multiple, but mostly benign, paragangliomas and phaeochromocytomas. SDHB mutations are associated with a higher proportion of malignant lesions and a relatively greater risk of phaeochromocytoma (8). SDHC mutations appear to be uncommon (4). Most lesions in HLRCC patients are benign smooth muscle tumours of the uterus (fibroids) or skin. Leiomyomsarcomas appear to occur in a small number of HLRCC families (9) and there is an established increased risk of renal cell carcinomas, of either type II papillary or collecting duct morphology and aggressive behaviour (10–13).
Despite the related roles of the SDH and FH proteins, the tumour spectra in HPGL and HLRCC have only a limited resemblance, and the underlying mechanism of tumorigenesis may not be the same in both syndromes. FH is a homotetramer which catalyses the hydration of fumarate to form malate. SDH, in contrast, consists of four separately encoded subunits (A–D) which catalyse the oxidative dehydrogenation of succinate and, as part of complex II of the electron transport chain, couple this to the reduction of ubiquinone (succinate–ubiquinone oxidoreductase). When succinate is oxidized to fumarate, two hydrogen atoms are removed from succinate and transferred to FAD, reducing it to FADH2 on the A subunit of SDH (SDHA). Electrons from FADH2 are sequentially transferred through three iron–sulphur centres in SDHB to the ubiquinone site associated with SDHC and SDHD, embedded in the mitochondrial inner membrane. It has even been proposed that the modes of tumorigenesis in SDHB and SDHD mutant may differ, because SDHD mutations are more likely to generate reactive oxygen species (ROSs) (14).
Several mechanisms have been proposed to account for the apparent paradox (15) of deficient energy production leading to tumorigenesis. These hypotheses include: (i) aberrant inactivation of hypoxia pathways (pseudo-hypoxic drive); (ii) increase in ROSs, leading to a stress response and/or hypermutation; (iii) defective apoptotic mechanisms resulting from primary mitochondrial dysfunction and (iv) a poorly defined mechanism of anabolic drive resulting from the build-up of glycolytic intermediates (reviewed in 16,17). The hypothesis that pseudo-hypoxic signalling drives tumorigenesis in HLRCC and HPGL is favoured by a comparison with the von Hippel–Lindau (VHL) syndrome. Mutations in VHL cause stabilization of the subunits of hypoxia-inducible factor (HIF) (18), which initiates the transcription of genes coding for pro-angiogenic proteins such as VEGF (19). In normoxia, HIF1α is usually hydroxylated at critical proline residues by one of at least three enzymes and thus targeted for degradation by an E3-ubiquitin ligase complex which involves the VHL protein. There is an overlap of phenotypes in HPGL and VHL syndromes, as individuals in both syndromes may develop highly vascular phaeochromocytomas (20,21). Clear cell renal carcinomas are a feature of VHL disease (22) and recently have been reported in a HPGL family (23).
The recent literature has provided evidence for a common mechanism of tumorigenesis in HPGL and HLRCC by reporting the up-regulation of hypoxia-responsive genes in tumours from both syndromes. Four tumours (three paragangliomas and one extra-adrenal phaeochromocytoma) from a family with an SDHD R22X mutation showed expression of vascular endothelial growth factor (VEGF) and endothelial PAS domain protein 1 (EPAS1/HIF2α) mRNAs at higher levels than that in sporadic phaeochromocytomas (24). HIF1α protein, generally regarded as the central mediator of the hypoxic response, was present at moderate levels in each of the four tumours, although its level in the sporadic lesions and in normal tissues was not determined. A single phaeochromocytoma from a patient with an R46Q SDHB mutation showed high expression of EPAS1 and VEGF mRNAs, but HIF1α was not studied (25). In a set of patients with germline FH mutations, uterine leiomyomas showed an increase in microvessel density, up-regulation of the VEGF and down-regulation of the angiogenesis inhibitor TSP1 in comparison with their sporadic counterparts and normal myometrium (26). Recently, support has been provided using an in vitro system for dysfunction of the Krebs cycle leading to the stabilization of HIF (27). Knockdown of SDHD resulted in the accumulation of succinate and inhibition of HIF-prolyl hydroxylase (PHD) activity, without the accumulation of free radicals.
We have investigated whether bi-allelic mutation of FH leads to accumulation of fumarate and/or succinate in cells from patients with inherited fumarase deficiency and in HLRCC tumours. We have also investigated FH-deficient cells for evidence of increased ROSs and oxidative stress. We then tested a panel of HLRCC tumours (fibroids and renal cancers) for accumulation of HIF1α and for associated changes in HIF-responsive genes such as VEGF. In addition, we have analysed tumours from HPGL patients and compared the findings with those from the HLRCC tumours. Our results are important for understanding the role of pseudo-hypoxic signalling in the pathogenesis of tumours in both HLRCC and HPGL.
RESULTS
Increased levels of succinate and/or fumarate in FH-deficient cells and HLRCC tumours
We initially used metabolomic profiling to study the effects of FH mutations on succinate and fumarate accumulation by analysing skin fibroblasts from four patients suffering from the recessive disorder fumarase deficiency. The enzyme activities and bi-allelic FH mutations in these patients had been previously determined (Table 1). As a control, we used six skin fibroblast cell lines from individuals with no apparent disease and no FH mutations. The FH-deficient cell lines had significantly higher levels (Table 2) of succinate (mean=59.4 µmol/g protein, median=39.8, range=32–125.9) than the normal cell lines (mean=17.4, median=14.5, range=9.9–29.8). The succinate:alpha-ketoglutarate ratio was also increased in all of the FH-deficient cells (data not shown); the method used was not specifically able to detect fumarate in these samples. We then tested for accumulation of succinate and fumarate in four HLRCC fibroids and in two normal myometrium samples, one from a HLRCC case and one from a non-HLRCC patient. Each tumour came from a patient with a known germline FH mutation and had acquired a ‘second hit’ by allelic loss. All of the tumour samples showed greatly elevated levels of both succinate and fumarate (Table 3). Normal myometrium samples from the HLRCC patient and the non-HLRCC patient showed similar levels of each metabolite. The combined data suggested that bi-allelic FH mutations in the HLRCC fibroids severely reduced the conversion of fumarate to malate, resulting in the blockage of the Krebs cycle and accumulation of fumarate and, consequently, succinate. The succinate:fumarate ratio was greater than one in the myometria, but typically 0.1–0.2 in the tumours (Table 3).
FH-deficient cells do not show notably increased oxidative stress and retain activity of mitochondrial complexes I, II–III and IV
The superoxide dismutase (SOD) assay was used on FH-deficient fibroblasts (n=2) and control fibroblasts (n=6) (Table 2). The former showed mean SOD levels of 44.4 mU/mg protein (median=44.4, range=38.7–50.1) and the latter showed very similar mean SOD levels of 38.2 mU/mg (median=38.0, range=31.3–47.3). A glutathione assay supported the absence of oxidative stress; the FH-deficient lines had a mean reduced glutathione concentration of 5.52 nmol/mg protein (median=5.52, range=3.30–7.74) and the control lines had a mean of 4.04 (median=3.44, range=1.72–7.34) (Table 2). The two FH-deficient lines were then tested to determine whether levels of the respiratory chain complexes and/or their abilities to transfer electrons were abnormal when compared with the six controls. There was no evidence that the levels of functional electron transport chain proteins were lower in the FH-deficient cells than that in controls (see Table 2 for complex I). Complex II–III activity was 0.096 and 0.010 in the two FH-deficient lines (reference range 0.070–0.243). Complex IV activity was 0.014 in one FH-deficient line when compared with a reference range of 0.007–0.036 and increased in the other at 0.120.
HIF1α expression in HLRCC tumours
We analysed 10 paraffin-embedded uterine fibroids, one frozen HLRCC papillary renal cell cancer and two paraffin-embedded HLRCC collecting duct cancers for the expression of nuclear HIF1α using immunohistochemistry. HIF1α staining was moderate in all of the fibroids studied, although weak/moderate staining was also observed in the surrounding myometrium. We had previously shown increased expression of VEGF in these lesions, associated with a high density of microvessels (26). In each of the three renal cancers, very strong HIF1α staining was observed in each tumour and staining was much weaker in the stromal tissue (Fig. 1). All of these lesions were found to be highly vascular. We confirmed the HIF1α over-expression by western blotting in the frozen renal tumour and normal kidney tissue. The 120 kDa band corresponding to HIF1α was consistently only present in the tumour (Fig. 2), thus verifying the results from the immunohistochemical analysis.
Identification of novel SDHB mutations and allelic loss
We screened constitutional DNA from six patients from whom we had frozen paragangliomas for mutations in SDHB, -C and -D. We found two novel germline variants in three of the patients. Both were missense mutations in exon 4 of SDHB. Patients 2 and 6 had a 380T>G change resulting in I127S and patient 4 had a 298T>C resulting in S100P. On array CGH, the two tumours from patients 2 and 6 had deletion of the whole of chromosome 1p (SDHB maps to 1p36.13) and fourth patient tumour had lost all of chromosome 1 (data not shown). Both of these mutations are likely to be pathogenic. They reside in residues which are entirely conserved throughout all eukaryotes and most prokaryotes, and occur in the iron–sulphur cluster binding domain of SDHB. S100P results in the substitution of a polar for a hydrophobic residue, which may result in impaired interactions between the SDHB and the other subunits of SDH. I127S results in a hydrophobic to polar residue change and may affect interactions within SDHB. Both mutations potentially affect the ability of SDHB to bind iron–sulphur clusters and neither was present in a set of 96 control individuals. We additionally screened constitutional DNA from the 19 patients with fixed, archival paragangliomas or phaeochromocytomas and we found several mutations previously reported in the literature in both SDHB (five paragangliomas and four phaeochromocytomas) and SDHD (four paragangliomas) (16).
Increased succinate in HPGL tumours
The six frozen paragangliomas were analysed using metabolomic profiling in order to determine the succinate and fumarate concentrations. Tumours from patients 2 and 6 with germline SDHB mutations had a grossly elevated concentration of succinate when compared with the three tumours from the non-HPGL patients (Table 4); the succinate:fumarate ratio was increased and positive in these lesions. One SDHB tumour (patient 4) did not show elevated succinate. These data showed that the SDHB mutations in the HPGL tumours can cause accumulation of succinate by impairing enzyme activity. The reason for the failure to detect elevated succinate in one tumour is unclear. It remains possible that S100P is a non-pathogenic change, but an increased succinate : fumarate ratio was seen in this tumour.
Paragangliomas from patients with germline SDHB and -D mutations show over-expression of HIF1α
We analysed all tumours from HPGL patients (comprising 12 paragangliomas and four phaeochromocytomas), including those used for metabolomic analysis, to test for hypoxic signalling. Strong nuclear expression of HIF1α was observed in all tumours studied (Fig. 3). Using in situ hybridization, we found VEGF expression to be high in all HPGL tumours when compared with the stroma and the surrounding tissues (Fig. 3). The expression pattern correlated with that of HIF1α. Evidence of the resulting angiogenesis was shown by the CD34 staining, which revealed a high density of microvessels throughout the tumours (data not shown).
DISCUSSION
Our results have demonstrated that bi-allelic FH mutations, whether in HLRCC tumours or in cells from fumarase deficiency patients, result in raised levels of both fumarate and succinate. Bi-allelic SDHB mutations predominantly cause raised levels of succinate. We have additionally shown that benign and malignant tumours in the HLRCC syndrome exhibit accumulation of the HIF1α protein, the central signalling molecule in the hypoxia pathway. HIF1α also accumulates in HPGL tumours, which result from germline SDHB mutations, adding to the existing data showing HIF1α expression in SDHD-mutant tumours. The raised levels of HIF1α result in increased expression of HIF-target genes, such as VEGF, and in angiogenesis.
Our evidence that over-expression of HIF1α is pathological, rather than a physiological response to tumour hypoxia, is 2-fold. First, we have observed no spatial association in leiomyomas, renal cancers, paragangliomas or phaeochromocytomas between vessel density as assessed by CD34 expression and HIF1α expression (data not shown). Thus, HIF1α expression is not simply caused by poor vasculature and consequent hypoxia. Secondly, we have found here that HIF1α levels are higher in HLRCC fibroids than in surrounding myometrium, and we have previously found that the expression levels of HIF1α targets, such as VEGF, TSP1 and BNIP1, are higher in HLRCC fibroids than in sporadic leiomyomas. However, microvessel density is highest in the HLRCC leiomyomas (26). If HIF1α expression were to be secondary to hypoxia, we would actually expect lower levels of its own expression and those of its target mRNAs in the more vascular HLRCC tumours.
Selak et al. (27) tested a model of tumorigenesis in which raised levels of succinate caused increased HIF1α expression in vitro. Our data provide evidence that raised succinate causes increased HIF1α expression in vivo. Selak et al. (27) showed that the mechanism for these findings may be inhibition by succinate of HIF PHDs, which rely for their activity on conversion of α-ketoglutarate to succinate. Dicarboxylic acid translocators in the mitochondrial inner membrane and voltage-dependent anion channels in the outer membrane allow succinate to move freely between the mitochondria and the cytosol where the HIF1α PHDs localize. Although physiological levels of succinate are difficult to achieve in vitro, our results show that succinate accumulates in HPGL and HLRCC tumours in vivo. It is not necessary for raised succinate to ‘signal’ the absence of Krebs cycle activity to the cell, in some sort of adaptive fashion; the build-up of succinate may merely prevent the forward HIF1α hydroxylation reaction. Alternative models of pseudo-hypoxic drive in tumorigenesis postulate that absence of Krebs cycle activity truly signals a need for increased oxygen to the cell, although it is not clear why such a signal should be required in addition to the physiological signalling which occurs through the hydroxylation of HIF by molecular oxygen. Whatever the correct model of pseudo-hypoxic drive, it should be emphasized that neither our data nor those of Selak et al. (27) have provided evidence for an alternative model of tumorigenesis in HLRCC or HPGL based on increased levels of ROSs.
The basic mechanism of tumorigenesis in HPGL and HLRCC is therefore likely to be pseudo-hypoxic drive, just as it is in von Hippel–Lindau syndrome. The tumours in all of these syndromes share morphological features, such as high vessel density, and molecular features, including increased levels of HIF1α Succinate accumulates in both HLRCC and HPGL tumours. However, we cannot exclude some direct inhibition of PHDs by fumarate in HLRCC. Although the double bond between the two central atoms makes fumarate a more rigid molecule than succinate, the two metabolites are chemically very similar. Our data showed that HLRCC tumours accumulate lower levels of succinate and higher levels of fumarate than HPGL tumours. Moreover, there is evidence to show that other metabolites, such as pyruvate and oxaloacetate, can stabilize HIF1α (38). The question remains as to why there are different, but overlapping, tumour spectra in the HPGL, HLRCC and VHL syndromes. Although several alternative explanations may exist, we suggest that the specific genetic defect (in FH, SDHB, SDHC or SDHD) might cause different levels of HIF1α accumulation with particular effects on cells which require different physiological responses to hypoxia, leading to the tissue specificity of the lesions in each diseases.
MATERIALS AND METHODS
FH deficiency patients
Fibroblast cell lines from four patients with FH deficiency were studied (Table 1) alongside normal control lines.
Tumour samples
We obtained fresh-frozen tissue from: four HLRCC fibroids; one sample of HLRCC normal myometrium; one sample of non-HLRCC myometrium; one HLRCC type II papillary renal cell cancer; unpaired normal kidney tissue and six paragangliomas of initially unknown HPGL status. We obtained fixed, paraffin-embedded material from 12 HLRCC fibroids and two sporadic fibroids, most with adjacent normal tissue, plus two HLRCC collecting duct renal carcinomas. In addition, we obtained fixed material from four phaeochromocytomas (including one lung metastasis) and 15 paragangliomas. All samples were collected with full Research Ethics Committee approval.
Mutation screening
FH mutation screening had been undertaken previously (11,13). For the SDH genes, mutation screening was performed by direct sequencing of genomic DNA in forward and reverse orientations using the Applied Biosystems Big Dye terminator reaction kit and the 377 semi-automated DNA sequencer. Primer sequences were designed to encompass the coding region and splice sites of each exon for SDHB, SDHC and SDHD (available on request).
Array comparative genome hybridization
The array construction, hybridization and analysis were performed as described (28,29). In brief, the array was prepared from DOP-PCR amplified DNA from 3452 large insert genomic clones at an average spacing of ∼1 Mb throughout the genome. DNA from each of the tumours was labelled with Cy5-dCTP. DNA from a pool of normals was used as a control and labelled with Cy-3dCTP. After correcting for background, the log2 ratio of the fluorescence intensities of tumour (T) to normal (N) was calculated, after normalization to the remainder of the genome in that tumour. Log2T:N ratios of >3 SD were taken to indicate the gain and ratios of <−3 SD were scored as loss on the basis of the previous N:N hybridizations. Copy number changes of the sex chromosomes were not analysed.
Cell culture
Human skin fibroblasts were cultured in Hams F10 media with added bacterial growth inhibitors (Gibco) in a humidified incubator at 37°C and 10% CO2. Cultures were split using standard methods and grown to 70–80% confluence for metabolomic studies. Normal skin fibroblasts were obtained from the European collection of cell cultures (ECACC).
FH activity assay of skin fibroblasts
The functional FH assay was a modification of a previously described method (30). Briefly, the assay monitors the increase in absorbance at 340 nm due to formation of NADPH in a linked assay of cell sonicate, with a final reaction medium (10 mm fumarate, 25 mm HEPES–KOH buffer pH 7.5, 0.2 Uml−1 malic enzyme, 0.27 mm NADP, 4 mm MgCl2 and 5 mm KH2PO4). The assay was run at least twice for each sample and at two different protein concentrations, and the final activity was expressed as a percentage of simultaneous normal controls.
Metabolite MRS analysis
For the assessment of succinate and other metabolites, MRS analysis was performed on FH-deficient and control fibroblast lines. Medium was removed from each flask and the cells were washed twice with 10 ml of phosphate-buffered saline. Two millilitres of 6% perchloric acid (PCA) were added to each flask which was gently rocked for 2 min to ensure that all the cells were covered by the PCA. Cells were scraped into a polypropylene tube and placed on ice. An additional 1 ml of 6% PCA was used to wash the flask and then added to the tubes which were centrifuged at 10 000 r.p.m. (Beckman) for 15 minutes at 4°C. The supernatant was pipetted into a clean tube and neutralized to pH 7.0 using 1%, 10% KOH and 1% PCA. The protein content of the pellet was determined by the Bradford assay (Biorad). The neutralized solution was further centrifuged at 10 000 r.p.m. for 10 min at 4°C and the supernatant pipetted into a clean tube and frozen. For the 1H MRS studies, the neutralized cell extracts were freeze-dried, reconstituted in D2O and placed in 5 mm NMR tubes. 1H MR spectra were obtained using a Bruker 600 MHz spectrometer (pulse angle, 45° and repetition time, 3.5 s). The water resonance was suppressed by gated irradiation centred on the water frequency. Sodium 3-trimethylsilyl-2,2,3,3-tetradeuterpropionate (TSP) was added to the samples for chemical shift calibration and quantification. The pH was re-adjusted to pH 7 prior to 1H MRS.
Tumour metabolite analysis
Frozen tumour samples from HLRCC, HPGL and other patients were homogenized in 1 ml of extraction buffer [Tris 20 mm (pH 7.2), sucrose 250 mm, EGTA 2 mm, KCl 40 mm, bovine serum albumin 1 mg/ml]. Samples were then centrifuged at 4°C for 30 min at 16 000g and the supernatant removed. Organic acids were quantified after acidification and ethylacetate extraction using gas chromatography/mass spectrometry, according to a standard procedure (31). The scarcity of the tumour samples and the need to treat each tumour with the optimum technique meant that duplicate analysis was not possible.
Analysis of oxidative stress: SOD assay
FH-deficient and control fibroblast cell pellets were re-suspended in 50 mmol/l Tris buffer containing 0.5% Triton X-100, pH 8.0 and lysed by freeze-thawing. Lysates were centrifuged at 13 000g for 10 min at 4°C. Determination of SOD activity in the cell lysates was performed as previously described (32), the method based on the ability of SOD to inhibit the autoxidation of pyrogallol being adapted for use on the Cobas Fara Autoanalyser (Roche Diagnostic, Welwyn Garden City, UK) using purified SOD from bovine erythrocytes (Sigma, UK) to construct a standard curve for this assay. Lysate protein content was measured using a Pierce BCA protein kit (BCA assay, Pierce, IL, USA). All data were expressed as units of SOD activity (U) per milligram protein. SOD assays were performed in duplicate and protein assays in triplicate.
Analysis of oxidative stress: glutathione assay
Oxidative stress in FH-deficient and control cells was measured using a HPLC method to analyse the oxidation status of the antioxidant glutathione as previously described (33).
Measurement of mitochondrial respiratory enzymes
Activities of mitochondrial respiratory chain enzymes were measured in FH-deficient and control cells as previously described [complex I (NADH ubiquinone reductase) (34), complex II–III (succinate:cytochrome c reductase) (35) and complex IV (cytochrome oxidase) (36)]. All respiratory chain activities were expressed as a ratio to citrate synthase activity to allow for mitochondrial enrichment (37).
Immunohistochemistry: HIF-1α
HIF1α staining was carried out using the CSA system (DAKO), as recommended by the manufacturer. In brief, tumour sections (4 µm) from fixed, paraffin-embedded were de-paraffinized in xylene, rehydrated and pressure cooked in target retrieval solution (16 p.s.i. for 2 min). Sections were blocked sequentially with avidin, biotin, hydrogen peroxidase and serum-free protein and incubated with the primary antibody (Novus) for 25 min. Sections were then treated with anti-mouse link antibody, stepavidin–biotin complex, amplification reagent, streptavidin-peroxidase and positive staining visualized with the addition of substrate chromagen solution (2–3 min). Slides were counterstained with haematoxylin for 30 s. Omission of primary antibody and non-tumour cells in the same section were used as a negative control and a known HIF1α-positive renal cell cancer was used as a positive control. For frozen samples, no antigen retrieval method was used and the sections were fixed in ice-cold acetone for 5 min prior to blocking and subsequent staining. Nuclear expression was scored by two independent observers (PP, NW, with confirmation is a subset by RP) as absent, weak, moderate or strong according to the proportion of tumour nuclei stained (0, 0–20, 20–80 and >80%, respectively).
Immunohistochemistry: CD34
Tumour sections (4 µm) were deparaffinized in xylene and rehydrated, and endogenous peroxide was blocked with 3% H2O2 (BDH Laboratory Supplies). Antigen retrieval was carried out in a pressure cooker for 3 min at 16 p.s.i. in 0.01 m citrate buffer. The CD34 primary antibody (M7165, Dako) was applied at a 1:25 dilution for 1 h; and antibody–antigen binding was detected using biotinylated goat anti-mouse antibodies (BA-9200, Vector Laboratories) at a 1:300 dilution This was followed by addition of peroxidase-conjugated streptavidin–biotin complex (Vector Laboratories). Sites of peroxidase activity were demonstrated using 3,3′-diaminobenzidine (Sigma) for 4 min. Slides were counterstained with haematoxylin for 30 s. Omission of primary antibody was used for a negative control and a known positive renal cell cancer was used for a positive control.
In situ hybridization
In situ hybridization was carried out using 4 µm serial sections from the same formalin-fixed, paraffin-embedded specimens as used for immunohistochemistry. The VEGF probe was a gift from Andrew Lee. The plasmid pGEM-3 containing cDNA (∼204 bases), from a region of VEGF common to all four splice variants, was HindIII-linearized and transcribed using T7 RNA polymerase. For hybridization control, a β-actin probe (∼414 bases) was generated from DraI-linearized pSP73 containing clone hβA-10. [35S]UTP in situ hybridization was performed as described previously. In brief, the sections were de-waxed, taken to water through graded alcohols and digested with proteinase K (20 mg/ml) for 10 min at 37°C. The sections were hybridized to 1×106 c.p.m. of each probe overnight at 55°C. Excess and poorly hybridized probe was destroyed by digestion with RNase A, and stringency washes were carried out to reduce non-specific binding. Slides were dipped in photographic emulsion (Ilford K5) and exposed at 4°C for 10 days before development (Kodak D-19). Patterns of hybridization were studied under dark-field reflected light, and signal was scored as absent, weak, moderate, strong or very strong by semi-quantitative observation, by two independent observers (RP and PP).
Western blotting
Levels of HIF1α protein were characterized in duplicate using standard western techniques. Briefly, frozen tumour tissue was homogenized in lysis buffer [7 m urea, 10% glycerol, 10 mm Tris–HCl (pH 6.8), 1% sodium dodecyl sulphate (SDS), 5 mm dithiothreitol (DTT), 0.5 mm phenylmethyl sulfonyl fluoride (PMSF) with 1 mg/l aprotinin, pepstatin and leupeptin], sonicated for 15 s (amplitude=30) and centrifuged at 4°C for 20 min at 10 000g. The supernatant was assayed for protein concentration (Biorad), denatured and 50 µg of total protein electrophoresed through 7.5% acrylamide gel (50 mA), followed by charged transfer to PVDF membrane (Millipore). Tumour and normal samples were incubated with mAb anti-HIF1α (Transduction laboratories) and detected using enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech, Amersham, UK). GAPDH (ABCAM) and β-actin (Sigma) were used for loading controls. HIF1α protein levels were quantitated by densitometry relative to the β-actin control.
ACKNOWLEDGEMENTS
We are grateful to the Cancer Research UK, London Research Institute Equipment Park and Histopathology Service, the patients who participated in this study and their doctors and Stefano Mandriota for advice on the HIF1α immunohistochemistry.
Conflict of Interest statement. None declared.
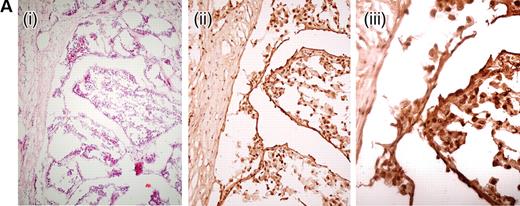
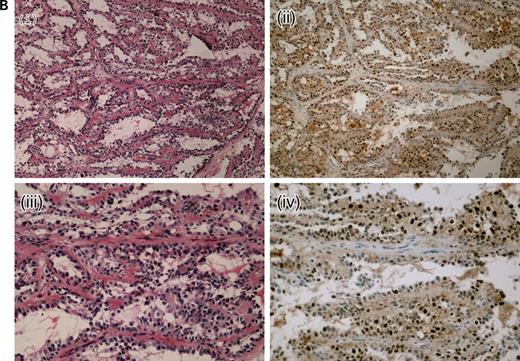
Figure 1. Representative HIF1α expression in HLRCC tumours by immunohistochemistry.(A) A frozen section of a HLRCC papillary type II renal tumour showing haemotoxylin and eosin (H&E) ×10 (i), HIF1α×10 (ii) and HIF1α×20 (iii). Even allowing for some background staining due to biotin levels in the kidney, there is strong nuclear expression of HIF1α.(B) A lymph node metastasis from a HLRCC collecting duct renal cancer showing H&E×10 (i), HIF1α×10 (ii), H&E×20 (iii) and HIF1α×20 (iv). There is strong HIF1α staining in the nuclei of tumour cells and negative staining in the stromal tissue, well-illustrated by the horizontal bands of stromal cells without HIF1α staining in (iv).
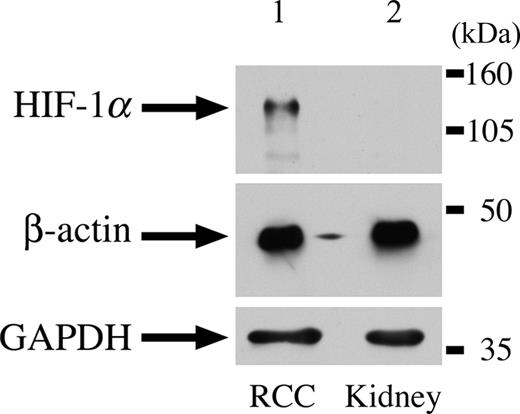
Figure 2. HIF1α expression in a HLRCC renal tumour by immunoblotting. Lane 1 represents total cellular lysate of type II papillary renal cell cancer from HLRCC patient. Lane 2 represents normal kidney. Bands corresponding to HIF-1α (120 kDa) and loading controls (GAPDH, 38 kDa and β-actin, 44 kDa) are arrowed.
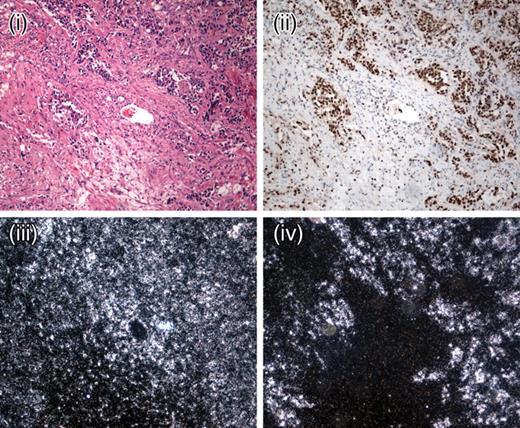
Figure 3. Representative expression of HIF1α and its target molecule VEGF in HPGL paragangliomas. SDHB HPGL paraganglioma (×10) showing H&E (i), HIF1α immunohistochemistry ×10 (ii), β-actin mRNA expression (iii) and VEGF mRNA expression (iv). Note the strong spatial association between the strong nuclear HIF1α staining and the VEGF expression in the tumour cells within ‘Zellballen’.
FH mutations and enzyme activity relative to controls in skin fibroblasts from FH-deficient patients
| Cell line | Mutation | Activity (%) |
|---|---|---|
| Fhdef1 | AAAins435/R58X | 1.9 |
| Fhdef2 | P131R/exon 2 splice variant | 13.0 |
| Fhdef3 | R190H/R190H | 2.4 |
| Fhdef4 | K187R/K187R | 6.0 |
| Cell line | Mutation | Activity (%) |
|---|---|---|
| Fhdef1 | AAAins435/R58X | 1.9 |
| Fhdef2 | P131R/exon 2 splice variant | 13.0 |
| Fhdef3 | R190H/R190H | 2.4 |
| Fhdef4 | K187R/K187R | 6.0 |
FH mutations and enzyme activity relative to controls in skin fibroblasts from FH-deficient patients
| Cell line | Mutation | Activity (%) |
|---|---|---|
| Fhdef1 | AAAins435/R58X | 1.9 |
| Fhdef2 | P131R/exon 2 splice variant | 13.0 |
| Fhdef3 | R190H/R190H | 2.4 |
| Fhdef4 | K187R/K187R | 6.0 |
| Cell line | Mutation | Activity (%) |
|---|---|---|
| Fhdef1 | AAAins435/R58X | 1.9 |
| Fhdef2 | P131R/exon 2 splice variant | 13.0 |
| Fhdef3 | R190H/R190H | 2.4 |
| Fhdef4 | K187R/K187R | 6.0 |
Levels of succinate, SOD and reduced glutathione and activities of complex I in FH-deficient cell lines and normal controls
| Sample | Succinate (µmol/g) | SOD (mU/mg) | Reduced gluthathione (nmol/mg) | Complex I |
|---|---|---|---|---|
| Control 1 | 17.1 | 31.3 | 4.90 | 0.952 |
| Control 2 | 9.9 | 38.3 | 3.44 | 0.267 |
| Control 3 | 11.4 | 37.7 | 1.72 | 0.764 |
| Control 4 | 24.5 | 35.7 | 2.81 | 0.486 |
| Control 5 | 29.8 | 38.7 | 7.34 | 0.783 |
| Control 6 | 11.8 | 47.3 | – | – |
| Fhdef1 | 32.6 | 38.7 | 7.74 | 1.72 |
| Fhdef2 | 125.9 | 50.1 | 3.30 | 0.981 |
| Fhdef3 | 32.0 | – | – | – |
| Fhdef4 | 47.0 | – | – | – |
| Sample | Succinate (µmol/g) | SOD (mU/mg) | Reduced gluthathione (nmol/mg) | Complex I |
|---|---|---|---|---|
| Control 1 | 17.1 | 31.3 | 4.90 | 0.952 |
| Control 2 | 9.9 | 38.3 | 3.44 | 0.267 |
| Control 3 | 11.4 | 37.7 | 1.72 | 0.764 |
| Control 4 | 24.5 | 35.7 | 2.81 | 0.486 |
| Control 5 | 29.8 | 38.7 | 7.34 | 0.783 |
| Control 6 | 11.8 | 47.3 | – | – |
| Fhdef1 | 32.6 | 38.7 | 7.74 | 1.72 |
| Fhdef2 | 125.9 | 50.1 | 3.30 | 0.981 |
| Fhdef3 | 32.0 | – | – | – |
| Fhdef4 | 47.0 | – | – | – |
Electron transport chain activities are shown relative to that of citrate synthase. –, not done. Formal statistical analysis is problematic, given the small number of samples and lack of information on the underlying distributions, although succinate levels are significantly higher in the FH-deficient patients (P=0.011, Wilcoxon test and P=0.05, t-test). No other significant differences between the two groups of patients exist (P>0.05, Wilcoxon and t-tests) data not shown).
Levels of succinate, SOD and reduced glutathione and activities of complex I in FH-deficient cell lines and normal controls
| Sample | Succinate (µmol/g) | SOD (mU/mg) | Reduced gluthathione (nmol/mg) | Complex I |
|---|---|---|---|---|
| Control 1 | 17.1 | 31.3 | 4.90 | 0.952 |
| Control 2 | 9.9 | 38.3 | 3.44 | 0.267 |
| Control 3 | 11.4 | 37.7 | 1.72 | 0.764 |
| Control 4 | 24.5 | 35.7 | 2.81 | 0.486 |
| Control 5 | 29.8 | 38.7 | 7.34 | 0.783 |
| Control 6 | 11.8 | 47.3 | – | – |
| Fhdef1 | 32.6 | 38.7 | 7.74 | 1.72 |
| Fhdef2 | 125.9 | 50.1 | 3.30 | 0.981 |
| Fhdef3 | 32.0 | – | – | – |
| Fhdef4 | 47.0 | – | – | – |
| Sample | Succinate (µmol/g) | SOD (mU/mg) | Reduced gluthathione (nmol/mg) | Complex I |
|---|---|---|---|---|
| Control 1 | 17.1 | 31.3 | 4.90 | 0.952 |
| Control 2 | 9.9 | 38.3 | 3.44 | 0.267 |
| Control 3 | 11.4 | 37.7 | 1.72 | 0.764 |
| Control 4 | 24.5 | 35.7 | 2.81 | 0.486 |
| Control 5 | 29.8 | 38.7 | 7.34 | 0.783 |
| Control 6 | 11.8 | 47.3 | – | – |
| Fhdef1 | 32.6 | 38.7 | 7.74 | 1.72 |
| Fhdef2 | 125.9 | 50.1 | 3.30 | 0.981 |
| Fhdef3 | 32.0 | – | – | – |
| Fhdef4 | 47.0 | – | – | – |
Electron transport chain activities are shown relative to that of citrate synthase. –, not done. Formal statistical analysis is problematic, given the small number of samples and lack of information on the underlying distributions, although succinate levels are significantly higher in the FH-deficient patients (P=0.011, Wilcoxon test and P=0.05, t-test). No other significant differences between the two groups of patients exist (P>0.05, Wilcoxon and t-tests) data not shown).
Succinate and fumarate levels (µmol/g protein) in HLRCC fibroids and control myometria
| Succinate | Fumarate | Succinate:fumarate | |
|---|---|---|---|
| Myometrium non-HLRCC | 6.0 | 2.0 | 3.0 |
| Myometrium HLRCC | 4.5 | 0.5 | 9.0 |
| HLRCC fibroid 1 | 88 | 458 | 0.19 |
| HLRCC fibroid 2 | 70 | 688 | 0.10 |
| HLRCC fibroid 3 | 98 | 646 | 0.15 |
| HLRCC fibroid 4 | 93 | 417 | 0.22 |
| Succinate | Fumarate | Succinate:fumarate | |
|---|---|---|---|
| Myometrium non-HLRCC | 6.0 | 2.0 | 3.0 |
| Myometrium HLRCC | 4.5 | 0.5 | 9.0 |
| HLRCC fibroid 1 | 88 | 458 | 0.19 |
| HLRCC fibroid 2 | 70 | 688 | 0.10 |
| HLRCC fibroid 3 | 98 | 646 | 0.15 |
| HLRCC fibroid 4 | 93 | 417 | 0.22 |
Although not significant by the Wilcoxon non-parametric test, a t-test shows both succinate (P=0.0009) and fumarate (P=0.0055) levels to be significantly higher in the tumour samples than the normal myometria.
Succinate and fumarate levels (µmol/g protein) in HLRCC fibroids and control myometria
| Succinate | Fumarate | Succinate:fumarate | |
|---|---|---|---|
| Myometrium non-HLRCC | 6.0 | 2.0 | 3.0 |
| Myometrium HLRCC | 4.5 | 0.5 | 9.0 |
| HLRCC fibroid 1 | 88 | 458 | 0.19 |
| HLRCC fibroid 2 | 70 | 688 | 0.10 |
| HLRCC fibroid 3 | 98 | 646 | 0.15 |
| HLRCC fibroid 4 | 93 | 417 | 0.22 |
| Succinate | Fumarate | Succinate:fumarate | |
|---|---|---|---|
| Myometrium non-HLRCC | 6.0 | 2.0 | 3.0 |
| Myometrium HLRCC | 4.5 | 0.5 | 9.0 |
| HLRCC fibroid 1 | 88 | 458 | 0.19 |
| HLRCC fibroid 2 | 70 | 688 | 0.10 |
| HLRCC fibroid 3 | 98 | 646 | 0.15 |
| HLRCC fibroid 4 | 93 | 417 | 0.22 |
Although not significant by the Wilcoxon non-parametric test, a t-test shows both succinate (P=0.0009) and fumarate (P=0.0055) levels to be significantly higher in the tumour samples than the normal myometria.
Succinate and fumarate levels (µmol/g protein) in HPGL and non-HPGL paragangliomas
| Tumour no. | Germline SDH mutation | Succinate | Fumarate | Succinate:fumarate |
|---|---|---|---|---|
| 1 | No | 49.7 | 12.7 | 3.9 |
| 2 | Yes | 364.5 | 17.8 | 20.5 |
| 3 | Yes | 43.2 | 12.6 | 3.4 |
| 4 | No | 32.3 | 3.0 | 10.8 |
| 5 | No | 40.9 | 7.0 | 5.8 |
| 6 | Yes | 517.0 | 13.9 | 37.2 |
| Tumour no. | Germline SDH mutation | Succinate | Fumarate | Succinate:fumarate |
|---|---|---|---|---|
| 1 | No | 49.7 | 12.7 | 3.9 |
| 2 | Yes | 364.5 | 17.8 | 20.5 |
| 3 | Yes | 43.2 | 12.6 | 3.4 |
| 4 | No | 32.3 | 3.0 | 10.8 |
| 5 | No | 40.9 | 7.0 | 5.8 |
| 6 | Yes | 517.0 | 13.9 | 37.2 |
Owing to sample 3, the differences between HPGL and other tumours are only just significantly different (P=0.045), despite the very large increases in succinate in tumours 2 and 6.
Succinate and fumarate levels (µmol/g protein) in HPGL and non-HPGL paragangliomas
| Tumour no. | Germline SDH mutation | Succinate | Fumarate | Succinate:fumarate |
|---|---|---|---|---|
| 1 | No | 49.7 | 12.7 | 3.9 |
| 2 | Yes | 364.5 | 17.8 | 20.5 |
| 3 | Yes | 43.2 | 12.6 | 3.4 |
| 4 | No | 32.3 | 3.0 | 10.8 |
| 5 | No | 40.9 | 7.0 | 5.8 |
| 6 | Yes | 517.0 | 13.9 | 37.2 |
| Tumour no. | Germline SDH mutation | Succinate | Fumarate | Succinate:fumarate |
|---|---|---|---|---|
| 1 | No | 49.7 | 12.7 | 3.9 |
| 2 | Yes | 364.5 | 17.8 | 20.5 |
| 3 | Yes | 43.2 | 12.6 | 3.4 |
| 4 | No | 32.3 | 3.0 | 10.8 |
| 5 | No | 40.9 | 7.0 | 5.8 |
| 6 | Yes | 517.0 | 13.9 | 37.2 |
Owing to sample 3, the differences between HPGL and other tumours are only just significantly different (P=0.045), despite the very large increases in succinate in tumours 2 and 6.
References
Baysal, B.E., Ferrell, R.E., Willett-Brozick, J.E., Lawrence, E.C., Myssiorek, D., Bosch, A., van der Mey, A., Taschner, P.E., Rubinstein, W.S., Myers, E.N. et al. (
Gimm, O., Armanios, M., Dziema, H., Neumann, H.P. and Eng, C. (
Niemann, S. and Muller, U. (
Astuti, D., Latif, F., Dallol, A., Dahia, P.L., Douglas, F., George, E., Skoldberg, F., Husebye, E.S., Eng, C. and Maher, E.R. (
Astuti, D., Douglas, F., Lennard, T.W., Aligianis, I.A., Woodward, E.R., Evans, D.G., Eng, C., Latif, F. and Maher, E.R. (
Tomlinson, I.P., Alam, N.A., Rowan, A.J., Barclay, E., Jaeger, E.E., Kelsell, D., Leigh, I., Gorman, P., Lamlum, H., Rahman, S. et al. (
Neumann, H.P., Pawlu, C., Peczkowska, M., Bausch, B., McWhinney, S.R., Muresan, M., Buchta, M., Franke, G., Klisch, J., Bley, T.A. et al. (
Kiuru, M., Lehtonen, R., Arola, J., Salovaara, R., Jarvinen, H., Aittomaki, K., Sjoberg, J., Visakorpi, T., Knuutila, S., Isola, J. et al. (
Kiuru, M. and Launonen, V. (
Alam, N.A., Rowan, A.J., Wortham, N.C., Pollard, P.J., Mitchell, M., Tyrer, J.P., Barclay, E., Calonje, E., Manek, S., Adams, S.J. et al. (
Toro, J.R., Nickerson, M.L., Wei, M.H., Warren, M.B., Glenn, G.M., Turner, M.L., Stewart, L., Duray, P., Tourre, O., Sharma, N. et al. (
Chan, I., Wong, T., Martinez-Mir, A., Christiano, A.M. and McGrath, J.A. (
Yankovskaya, V., Horsefield, R., Tornroth, S., Luna-Chavez, C., Miyoshi, H., Leger, C., Byrne, B., Cecchini, G. and Iwata, S. (
Eng, C., Kiuru, M., Fernandez, M.J. and Aaltonen, L.A. (
Pollard, P.J., Wortham, N.C. and Tomlinson, I.P.M. (
Maxwell, P.H., Wiesener, M.S., Chang, G.W., Clifford, S.C., Vaux, E.C., Cockman, M.E., Wykoff, C.C., Pugh, C.W., Maher, E.R. and Ratcliffe, P.J. (
Jones, A., Fujiyama, C., Blanche, C., Moore, J.W., Fuggle, S., Cranston, D., Bicknell, R. and Harris, A.L. (
Gimenez-Roqueplo, A.P., Favier, J., Rustin, P., Rieubland, C., Crespin, M., Nau, V., Khau Van Kien, P., Corvol, P., Plouin, P.F. and Jeunemaitre, X. (
Clifford, S.C. and Maher, E.R. (
Clifford, S.C., Prowse, A.H., Affara, N.A., Buys, C.H. and Maher, E.R. (
Vanharanta, S., Buchta, M., McWhinney, S.R., Virta, S.K., Peczkowska, M., Morrison, C.D., Lehtonen, R., Januszewicz, A., Jarvinen, H., Juhola, M. et al. (
Gimenez-Roqueplo, A.P., Favier, J., Rustin, P., Mourad, J.J., Plouin, P.F., Corvol, P., Rotig, A. and Jeunemaitre, X. (
Gimenez-Roqueplo, A.P., Favier, J., Rustin, P., Rieubland, C., Kerlan, V., Plouin, P.F., Rotig, A. and Jeunemaitre, X. (
Pollard, P., Wortham, N., Barclay, E., Alam, A., Elia, G., Manek, S., Poulsom, R. and Tomlinson, I. (
Selak, M.A., Armour, S.M., MacKenzie, E.D., Boulahbel, H., Watson, D.G., Mansfield, K.D., Pan, Y., Simon, M.C., Thompson, C.B. and Gottlieb, E. (
Douglas, E.J., Fiegler, H., Rowan, A., Halford, S., Bicknell, D.C., Bodmer, W., Tomlinson, I.P.M. and Carter, N.P. (
Fiegler, H., Carr, P., Douglas, E.J., Burford, D.C., Hunt, S., Scott, C.E., Smith, J., Vetrie, D., Gorman, P., Tomlinson, I.P.M. et al. (
Hatch, M.D. (
Sandstrom, B.E., Granstrom, M., Vezin, H., Bienvenu, P. and Marklund, S.L. (
Riederer, P., Sofic, E., Rausch, W.D., Schmidt, B., Reynolds, G.P., Jellinger, K. and Youdim, M.B. (
Darley-Usmar, V.M., Rickwood, D. and Wilson, M.T. (
King, T.S. (
Wharton, D.C. and Tzagoloff, A. (
Shephard, D. and Garland P.B. (

{kind=link}
{kind=link}
{kind=link}