




Abstract
Myopathy is a typical clinical finding among patients with the 3243A>G mutation in mitochondrial DNA (mtDNA), but the variability in such findings has not been properly established. We have previously determined the prevalence of patients with 3243A>G in a defined population in northern Finland and characterized a group of patients who represent a good approximation to a population-based cohort. We report here on examinations performed on patients belonging to this cohort in order to determine the frequency of myopathy and to evaluate the clinical, histological, ultrastructural and single fibre mtDNA variability in muscle involvement. Fifty patients with 3243A>G underwent a thorough structured interview and clinical examination. Muscle histology, ultrastructure and single fibre analysis were examined in a subset of patients. A clinical diagnosis of myopathy was made in 50% of cases [95% confidence interval (CI), 36–64] and abnormalities in muscle histology were found in 72% (95% CI, 55–86). Moderate limb weakness leading to functional impairment was the most common myopathic sign, but mild weakness, ptosis and external ophthalmoplegia could also be found. The presence of intramitochondrial crystals and cytochrome c oxidase (COX)-negative fibres and variation in mitochondrial size and shape were more common in the muscles of the myopathic patients. Longitudinal variations in mutation heteroplasmy were examined in single muscle fibres from two severely affected patients. Although the total variation in mutation heteroplasmy along four ragged red fibres (RRFs) was small, the mutation heteroplasmy in five 10 µm segments was clearly lower (median 68%, range 64–74%) than that in the neighbouring segments. There were also segments with deviant mutation load in histologically normal fibres in one patient. The highest incidence of myopathy was in the fifth decade of life, but, apart from age, no other clinical variables such as gender, muscle heteroplasmy, physical inactivity or diabetes were associated with an increased risk of myopathy. The clinical presentation of myopathy is highly variable in patients with 3243A>G.
Introduction
Clinical features are quite varied in patients harbouring the 3243A>G point mutation in the gene encoding tRNALeu(UUR) (MTTL1) in mitochondrial DNA (mtDNA), the most common cause of MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes) syndrome (Goto et al., 1990). The varied phenotypes are at least partly explained by the fact that the mutation is heteroplasmic, i.e. both the wild-type and the mutant genome is present in tissues. The proportion of the mutant genome may vary between tissues as a result of replicative segregation, and a biochemical defect in the respiratory chain ensues if this proportion exceeds a critical threshold (DiMauro and Schon, 2003). Therefore, patients with a high proportion of mutant mtDNA in their tissue are more likely to be affected.
Muscle involvement is common among patients with 3243A>G, and the main clinical manifestations include proximal limb weakness, ptosis and progressive external ophthalmoplegia (Moraes et al., 1993). The reported frequencies of myopathy among patients with 3243A>G have varied from 25% (Deschauer et al., 2001) to 61% (Hammans et al., 1995), and a meta-analysis has suggested a frequency of 53% (Chinnery et al., 1997). Morphological alterations are also common in the skeletal muscle of patients with mitochondrial myopathy including ragged red fibres (RRFs) that often stain negative for cytochrome c oxidase (COX), although these may also stain positive in the case of patients with 3243A>G. Ultrastructurally, there is an excess of abnormal subsarcolemmal mitochondria. No clear correlations have been found between various ultrastructural alterations and clinical phenotypes (Morgan-Hughes, 1994).
The proportion of mutant mtDNA is variable even among the cells in a given tissue, and such a mosaic pattern is most evident in muscle, where RRFs, COX-negative fibres and normal fibres are clearly distinct (Petruzzella et al., 1994). Furthermore, COX deficiency shows segmental distribution along the muscle fibres of patients with mitochondrial myopathies (Larsson and Oldfors, 2001). Single fibre analysis of muscle cross-sections from patients with 3243A>G has revealed differences in mutation load among the cells (Petruzzella et al., 1994), but the variations in the proportion of the mutant genome along individual muscle fibres are not known.
We have previously determined the prevalence of patients with 3243A>G in a defined population in northern Finland (Majamaa et al., 1998). The mutation was not uncommon, implying that mitochondrial diseases must be considered in the differential diagnosis of myopathies. Although a multitude of clinical and histopathological features of myopathy have been documented in patients with 3243A>G, the spectrum of these findings has been inadequately studied. We therefore set out to examine 50 patients with 3243A>G who had been ascertained in the population-based screening and thus constitute a good approximation to a population-based cohort. We determined the frequency of myopathy among these patients and evaluated the clinical, histological and ultrastructural variability of muscle involvement.
Patients and methods
Clinical examination
Fifty patients (18 men, 32 women) aged 16–70 years (45 ± 14 years) were clinically examined by the same neurologist. The patients harboured the 3243A>G mutation in their mtDNA and they were from 17 pedigrees that have previously been ascertained in the province of Northern Ostrobothnia, Finland, following an epidemiological survey (see Majamaa et al., 1998). One complete sibship from each pedigree and their mothers was intended to be included in the present study. The total number of adult members in these sibships was 102 subjects, 30 of whom were deceased and 33 siblings remained unexamined. The most common cause of non-participation was the fact that the person lived elsewhere in the country (n = 17). Six mothers were examined, eight had died before the study and three did not participate. In addition, five adult children of a female carrier were also included.
All subjects had had normal early development. Their current physical activity and susceptibility to exercise intolerance were determined by means of a structured interview. A composite Medical Research Council (MRC) score was used to assess muscle power (Katirji, 2002). Ten muscle functions were tested on each side, including arm abduction, elbow flexion, elbow extension, wrist extension, finger abduction, hand grip, hip flexion, knee extension, knee flexion and ankle dorsiflexion. Because MRC score 4 encompasses the majority of patients with muscle weakness, a modified scale with scores subdivided into quarters was used (Mendel and Florence, 1990). Normal muscle power would thus give a total score of 100. An MRC score ≤90 was considered to indicate moderate myopathy and 90 < MRC < 100 was considered to indicate mild myopathy. In addition, patients with extraocular muscle weakness with or without other cranial muscle weakness were considered myopathic. The time required to stand up from the supine position was measured, and stepping onto a 30 cm high footstool and gait were observed (Cwik and Brooke, 1996). The stepping test was defined as abnormal if hesitation, hip dip, jump or hand support were observed when stepping onto the footstool, and gait as abnormal if waddling or any ambulatory disturbances attributable to muscle weakness were observed. The assessment of the global clinical severity of the disease was based on this functional evaluation and was scored in terms of the modified Rankin scale (0–5). The age at onset in the patients with myopathy was determined on the basis of a history. If the history was inconclusive, patient charts were reviewed, and if this was inconclusive, the age at examination was regarded as the age at onset. These patients were symptomatic, but could not recall the onset of symptoms or had not sought medical care.
Venous blood lactate at rest (normal, <1.4 mmol/l) and serum creatine kinase (normal, <270 U/l for men and <150 U/l for women) was measured in 44 cases. Other causes of myopathy were excluded by means of a family history, neurological examination and laboratory tests. Mutation heteroplasmy in muscle was determined by restriction fragment analysis followed by correction for heteroduplex formation (Kobayashi et al., 1990; Shoffner et al., 1990). In short, a fragment of mtDNA was amplified in 30 cycles in the presence of [35S]dATP. The digested samples were electrophoresed through a 6% acrylamide gel, which was autoradiographed and the exposed film was analysed for band intensities with a Bioimage scanner and image processing apparatus (Millipore).
The study protocol has been approved by the Ethics Committee of Oulu University Hospital, and written informed consent was obtained from all the patients.
Muscle histochemistry and ultrastructural analyses
A muscle biopsy was taken under local anaesthesia from 36 patients (vastus lateralis of the quadriceps muscle, 27 patients; anterior tibial muscle, nine patients) including 14 probands in the 17 families and 22 secondary cases. The 14 non-biopsied patients included three probands and 11 secondary cases in eight different families and they were not considered for biopsy, because the diagnosis had already been verified by molecular methods. Frozen sections of muscle were stained for haematoxylin and eosin (HE), modified Gomori trichrome, NADH-tetrazolium reductase and ATPase after incubation at pH 9.4, 4.6 and 4.3, respectively. Myopathic findings such as variation in fibre size, central nuclei, reactive fibrosis and necrotic or basophilic muscle fibres were observed, as were fat infiltration and atrophic and inflammatory changes. RRFs were identified in sections stained with modified Gomori trichrome. RRFs were regarded as numerous if their proportion exceeded 5% of total muscle fibres, moderate if they were between 2 and 5% and few in number if the proportion was <2%. In addition, reactions for COX and succinate dehydrogenase (SDH) (Sciacco et al., 1994) were performed in muscle samples from 16 patients, who were examined after the implementation of this method in the laboratory. The proportions of COX-negative fibres and RRFs in each muscle specimen were determined by counting 200–500 muscle fibres.
Electron microscopy was performed on all the biopsy specimens except for two, which were considered to be inadequate for diagnosis. Fresh tissue was fixed in 4% formaldehyde/1% glutaraldehyde buffered to pH 7.4 with phosphate buffer and post-fixed in OsO4 and embedded in Epon LX-112 (Electron Microscopy Sciences, Fort-Washington, PA). Ultra-thin sections were contrasted with uranyl acetate and lead citrate and examined in a Philips LS instrument (Philips Export B.V., Eindhoven, The Netherlands). The findings were documented on photographs. Mild mitochondrial changes were diagnosed when only a slightly increased number of mitochondria or slight alterations in size and shape were seen. Severe mitochondrial changes were diagnosed when a markedly increased number of mitochondria or pronounced alterations in size and shape were seen. Intramitochondrial crystals were also observed; the type I crystals, referred to as ‘parking lot’ inclusions, located in the intracristal spaces and the type II crystals, usually located between the inner and outer mitochondrial membranes (Stadhouders et al., 1994), were each determined as either present or absent. Giant mitochondria, cristae alterations and fat were also estimated quantitatively, while lipofuscin and osmiophilic balls were assessed as being present or absent.
Isolation of single muscle fibres
Single muscle fibres were dissected out with a sharp tungsten needle (Oldfors et al., 1995). The proportion of mutant mtDNA was quantified in cross-sections from the 10 patients, including 251 histologically normal fibres (18–29 fibres per subject), eight COX-negative non-RRF fibres, 19 COX-positive RRFs and 13 COX-negative RRFs. In addition, we analysed 10 serial transverse cross-sections from adjacent fascicles in two severely affected patients. Two separate COX-positive RRFs were confidently identified in all the 10 sections, and the RRFs and all the histologically normal fibres adjacent to the RRFs were dissected out. The intervening space between the sections was 10 µm and the total length of the specimen analysed was 200 µm. A total of 35 RRFs and 206 histologically normal fibres adjacent to RRFs were successfully dissected out and analysed from the two patients.
Single fibre mtDNA analysis
To analyse the proportion of mutant and wild-type mtDNA, each isolated muscle fibre segment was incubated for 10 min at 94°C in 5 µl of a solution of 200 mM KOH and 50 mM dithiothreitol and neutralized. A portion of the lysate was then taken for DNA amplification. A fragment spanning nucleotides 3187 and 3410 of mtDNA was amplified by polymerase chain reaction (PCR) in 35 cycles with denaturation at 94°C for 60 s, primer annealing at 49°C for 60 s and primer extension at 72°C for 60 s in the presence of 1.0 U of DNA polymerase (Finnzymes, Helsinki, Finland). For quantification of the percentage of mutation, 10 pmol 6-FAM (6-carboxyfluorescein)-labelled forward primer and 1.0 U of DNA polymerase were added to each PCR before the last cycle. The amplified fragments were then digested with 10 U of ApaI (New England Biolabs, Beverly, MA) overnight at 37°C. The digestion products were electrophoresed through a 4% polyacrylamide gel using a model 377 DNA sequencer (Applied Biosystems, Foster City, CA) and analysed with the Genotyper version 2.0 software. The proportions of mutant and wild-type mtDNAs were calculated from the peak areas of cleaved and uncleaved mtDNAs (Moslemi et al., 1998). A standard curve, constructed by mixing different proportions of cloned mutant and wild-type mtDNAs, was linear. The SD of the measurements was estimated to 0.6%. The heteroplasmy of each fibre was determined twice, and analyses were accepted if the difference between the two measurements was ≤2 percentage units.
Statistical analyses
The Mann–Whitney U test was used to detect differences between two groups. Bonferroni correction for multiple comparisons was applied by multiplying the observed P value by the number of tests, and P values <0.05 after correction were considered significant. The P values shown are the uncorrected ones.
Results
Myopathy phenotype
Twenty-five patients (50%) presented with myopathic findings (Table 1). Thirteen of these patients had myopathy with moderate proximal limb weakness, and eight patients (16%) were found with mild proximal limb weakness. Four (8%) had ptosis or ophthalmoplegia. Three patients had experienced muscle fatigue, but no cardiopulmonary symptoms, after a modest exercise, suggesting exercise intolerance (Table 1). Fourteen patients of the present study have been examined previously for muscle CT (Kärppä et al., 2004). Muscle CT appeared to be more often abnormal in patients with moderate limb weakness than in patients with mild limb weakness (Table 1).
Clinical and demographic features of the 25 patients with myopathy
| Patient | Gender/age (years) | Clinical features | HP (%) | Myopathy characteristics | Decade of mp | MRC | Rankin score | CK (U/l) | RRF | CT | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patients with moderate limb weakness | ||||||||||||||||||||
| 2 | F/68 | G, H, L, S | 53 | Moderate proximal, mild waddling, myalgia | 5 | 82.5 | 2 | 177 | 1 | 2 | ||||||||||
| 7 | F/55 | G, HA, L, S | 67 | Moderate proximal, facial, asthenic | 5 | 82 | 2 | 144 | 1 | 3 | ||||||||||
| 11 | F/40 | B, C, E, G, H, L, N, R | 73 | Moderate proximal, waddling, asthenic | 3 | 85 | 3 | n.d. | 3 | n.d. | ||||||||||
| 13 | M/61 | G | 64 | Moderate proximal | 7 | 80 | 3 | 74 | 3 | n.d. | ||||||||||
| 15 | M/53 | A, D, G, L, N, S | 72 | Moderate general, facial, palatal, waddling | 4 | 82.5 | 3 | 358 | 0 | 3 | ||||||||||
| 16 | M/63 | C, D, G, H, L, N, S | 70 | Moderate proximal, ptosis | 5 | 89.5 | 3 | 188 | 3 | 2 | ||||||||||
| 18 | F/17 | E, G,HA, S | n.d. | Moderate general, facial | 2 | 81.5 | 2 | 57 | n.d. | 1 | ||||||||||
| 28 | F/65 | A, C, D, G, HE, L, N, S | 80 | Moderate general, facial, palatal, waddling, asthenic | 5 | 84 | 3 | 144 | 1 | n.d. | ||||||||||
| 29 | F/33 | D, E, G, HA, L, S | 86 | Moderate general, asthenic | 2 | 80 | 3 | 187 | 2 | n.d. | ||||||||||
| 30 | M/43 | C, S, N | 43 | Moderate proximal, facial, waddling, asthenic | 2 | 88.5 | 2 | n.d. | 0 | n.d. | ||||||||||
| 35 | M/65 | C, D, G, H, L, N, S | 57 | Moderate general, walking with assistance, asthenic | 5 | 89 | 4 | 122 | 1 | n.d. | ||||||||||
| 40 | F/62 | C, G, H, O, S | 66 | Moderate general, facial, waddling | 5 | 80.5 | 2 | 209 | 0 | 2 | ||||||||||
| 45 | M/54 | A, D, L, N, O, S | 74 | Moderate general, ptosis, facial, waddling | 5 | 82 | 3 | n.d. | 2 | n.d. | ||||||||||
| Patients with mild limb weakness | ||||||||||||||||||||
| 4 | F/48 | D, G, L, S, V | 75 | Mild, only thighs | 5 | 97.5 | 2 | 214 | 1 | 0 | ||||||||||
| 6 | F/20 | E, G, HA, L, R, S, SE | n.d. | Mild proximal | 2 | 96 | 2 | 235 | n.d. | 3 | ||||||||||
| 9 | M/16 | C, G, H, S | 76 | Mild proximal | 2 | 95 | 2 | 134 | 1 | 0 | ||||||||||
| 12 | M/49 | L | n.d. | Mild proximal | 5 | 95 | 1 | 225 | n.d. | 1 | ||||||||||
| 19 | F/48 | S | 74 | EI, myalgia, mild proximal | 5 | 98 | 1 | 109 | 0 | 0 | ||||||||||
| 36 | F/70 | D, G, S | n.d. | Mild proximal | 7 | 96 | 1 | 93 | n.d. | 3 | ||||||||||
| 43 | F/52 | D, G, H, L | 78 | Mild proximal | 6 | 96 | 1 | 133 | 1 | 2 | ||||||||||
| 46 | M/57 | C, G, HE | n.d. | Mild proximal, lower limbs | 6 | 97 | 2 | n.d. | n.d. | n.d. | ||||||||||
| Patients with ptosis | ||||||||||||||||||||
| 5 | F/44 | A, D, H, L, O, S | 67 | Ptosis, EI | 5 | 100 | 1 | 206 | 1 | n.d. | ||||||||||
| 8 | F/38 | C, G, H, S | 48 | Ptosis | 4 | 100 | 1 | 82 | 0 | n.d. | ||||||||||
| 10 | F/38 | G | 48 | Ptosis | 4 | 100 | 1 | 106 | 0 | n.d. | ||||||||||
| 24 | M/57 | D, G, H | 73 | Ptosis, EI, myalgia | 6 | 100 | 2 | 221 | 1 | 0 | ||||||||||
| Patient | Gender/age (years) | Clinical features | HP (%) | Myopathy characteristics | Decade of mp | MRC | Rankin score | CK (U/l) | RRF | CT | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patients with moderate limb weakness | ||||||||||||||||||||
| 2 | F/68 | G, H, L, S | 53 | Moderate proximal, mild waddling, myalgia | 5 | 82.5 | 2 | 177 | 1 | 2 | ||||||||||
| 7 | F/55 | G, HA, L, S | 67 | Moderate proximal, facial, asthenic | 5 | 82 | 2 | 144 | 1 | 3 | ||||||||||
| 11 | F/40 | B, C, E, G, H, L, N, R | 73 | Moderate proximal, waddling, asthenic | 3 | 85 | 3 | n.d. | 3 | n.d. | ||||||||||
| 13 | M/61 | G | 64 | Moderate proximal | 7 | 80 | 3 | 74 | 3 | n.d. | ||||||||||
| 15 | M/53 | A, D, G, L, N, S | 72 | Moderate general, facial, palatal, waddling | 4 | 82.5 | 3 | 358 | 0 | 3 | ||||||||||
| 16 | M/63 | C, D, G, H, L, N, S | 70 | Moderate proximal, ptosis | 5 | 89.5 | 3 | 188 | 3 | 2 | ||||||||||
| 18 | F/17 | E, G,HA, S | n.d. | Moderate general, facial | 2 | 81.5 | 2 | 57 | n.d. | 1 | ||||||||||
| 28 | F/65 | A, C, D, G, HE, L, N, S | 80 | Moderate general, facial, palatal, waddling, asthenic | 5 | 84 | 3 | 144 | 1 | n.d. | ||||||||||
| 29 | F/33 | D, E, G, HA, L, S | 86 | Moderate general, asthenic | 2 | 80 | 3 | 187 | 2 | n.d. | ||||||||||
| 30 | M/43 | C, S, N | 43 | Moderate proximal, facial, waddling, asthenic | 2 | 88.5 | 2 | n.d. | 0 | n.d. | ||||||||||
| 35 | M/65 | C, D, G, H, L, N, S | 57 | Moderate general, walking with assistance, asthenic | 5 | 89 | 4 | 122 | 1 | n.d. | ||||||||||
| 40 | F/62 | C, G, H, O, S | 66 | Moderate general, facial, waddling | 5 | 80.5 | 2 | 209 | 0 | 2 | ||||||||||
| 45 | M/54 | A, D, L, N, O, S | 74 | Moderate general, ptosis, facial, waddling | 5 | 82 | 3 | n.d. | 2 | n.d. | ||||||||||
| Patients with mild limb weakness | ||||||||||||||||||||
| 4 | F/48 | D, G, L, S, V | 75 | Mild, only thighs | 5 | 97.5 | 2 | 214 | 1 | 0 | ||||||||||
| 6 | F/20 | E, G, HA, L, R, S, SE | n.d. | Mild proximal | 2 | 96 | 2 | 235 | n.d. | 3 | ||||||||||
| 9 | M/16 | C, G, H, S | 76 | Mild proximal | 2 | 95 | 2 | 134 | 1 | 0 | ||||||||||
| 12 | M/49 | L | n.d. | Mild proximal | 5 | 95 | 1 | 225 | n.d. | 1 | ||||||||||
| 19 | F/48 | S | 74 | EI, myalgia, mild proximal | 5 | 98 | 1 | 109 | 0 | 0 | ||||||||||
| 36 | F/70 | D, G, S | n.d. | Mild proximal | 7 | 96 | 1 | 93 | n.d. | 3 | ||||||||||
| 43 | F/52 | D, G, H, L | 78 | Mild proximal | 6 | 96 | 1 | 133 | 1 | 2 | ||||||||||
| 46 | M/57 | C, G, HE | n.d. | Mild proximal, lower limbs | 6 | 97 | 2 | n.d. | n.d. | n.d. | ||||||||||
| Patients with ptosis | ||||||||||||||||||||
| 5 | F/44 | A, D, H, L, O, S | 67 | Ptosis, EI | 5 | 100 | 1 | 206 | 1 | n.d. | ||||||||||
| 8 | F/38 | C, G, H, S | 48 | Ptosis | 4 | 100 | 1 | 82 | 0 | n.d. | ||||||||||
| 10 | F/38 | G | 48 | Ptosis | 4 | 100 | 1 | 106 | 0 | n.d. | ||||||||||
| 24 | M/57 | D, G, H | 73 | Ptosis, EI, myalgia | 6 | 100 | 2 | 221 | 1 | 0 | ||||||||||
HP = mutation heteroplasmy in muscle; Decade of mp = decade of life, during which symptoms of myopathy first occurred; MRC = composite Medical Research Council score; Rankin score, 1 = no significant disability, 2 = slight disability, 3 = moderate disability, 4 = moderately severe disability; CK = creatine kinase; RRF = ragged red fibres (0 = none, 1 = few, 2 = moderate number, 3 = many); CT = muscle computed tomography (0 = normal, 1 = mild, 2 = moderate, 3 = severe changes); F = female; M = male; A = ataxia; B = basal ganglia calcifications; C = cognitive decline; D = diabetes mellitus; E = epilepsy; EI = exercise intolerance; G = short stature; H = cardiac hypertrophy; HA = occasional headache; HE = hepatopathy; L = lactic acidosis; N = peripheral neuropathy; O = ophthalmoplegia; R = pigment retinopathy; S = sensorineural hearing impairment; SE = stroke-like episodes; V = vitiligo; n.d. = not determined.
Clinical and demographic features of the 25 patients with myopathy
| Patient | Gender/age (years) | Clinical features | HP (%) | Myopathy characteristics | Decade of mp | MRC | Rankin score | CK (U/l) | RRF | CT | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patients with moderate limb weakness | ||||||||||||||||||||
| 2 | F/68 | G, H, L, S | 53 | Moderate proximal, mild waddling, myalgia | 5 | 82.5 | 2 | 177 | 1 | 2 | ||||||||||
| 7 | F/55 | G, HA, L, S | 67 | Moderate proximal, facial, asthenic | 5 | 82 | 2 | 144 | 1 | 3 | ||||||||||
| 11 | F/40 | B, C, E, G, H, L, N, R | 73 | Moderate proximal, waddling, asthenic | 3 | 85 | 3 | n.d. | 3 | n.d. | ||||||||||
| 13 | M/61 | G | 64 | Moderate proximal | 7 | 80 | 3 | 74 | 3 | n.d. | ||||||||||
| 15 | M/53 | A, D, G, L, N, S | 72 | Moderate general, facial, palatal, waddling | 4 | 82.5 | 3 | 358 | 0 | 3 | ||||||||||
| 16 | M/63 | C, D, G, H, L, N, S | 70 | Moderate proximal, ptosis | 5 | 89.5 | 3 | 188 | 3 | 2 | ||||||||||
| 18 | F/17 | E, G,HA, S | n.d. | Moderate general, facial | 2 | 81.5 | 2 | 57 | n.d. | 1 | ||||||||||
| 28 | F/65 | A, C, D, G, HE, L, N, S | 80 | Moderate general, facial, palatal, waddling, asthenic | 5 | 84 | 3 | 144 | 1 | n.d. | ||||||||||
| 29 | F/33 | D, E, G, HA, L, S | 86 | Moderate general, asthenic | 2 | 80 | 3 | 187 | 2 | n.d. | ||||||||||
| 30 | M/43 | C, S, N | 43 | Moderate proximal, facial, waddling, asthenic | 2 | 88.5 | 2 | n.d. | 0 | n.d. | ||||||||||
| 35 | M/65 | C, D, G, H, L, N, S | 57 | Moderate general, walking with assistance, asthenic | 5 | 89 | 4 | 122 | 1 | n.d. | ||||||||||
| 40 | F/62 | C, G, H, O, S | 66 | Moderate general, facial, waddling | 5 | 80.5 | 2 | 209 | 0 | 2 | ||||||||||
| 45 | M/54 | A, D, L, N, O, S | 74 | Moderate general, ptosis, facial, waddling | 5 | 82 | 3 | n.d. | 2 | n.d. | ||||||||||
| Patients with mild limb weakness | ||||||||||||||||||||
| 4 | F/48 | D, G, L, S, V | 75 | Mild, only thighs | 5 | 97.5 | 2 | 214 | 1 | 0 | ||||||||||
| 6 | F/20 | E, G, HA, L, R, S, SE | n.d. | Mild proximal | 2 | 96 | 2 | 235 | n.d. | 3 | ||||||||||
| 9 | M/16 | C, G, H, S | 76 | Mild proximal | 2 | 95 | 2 | 134 | 1 | 0 | ||||||||||
| 12 | M/49 | L | n.d. | Mild proximal | 5 | 95 | 1 | 225 | n.d. | 1 | ||||||||||
| 19 | F/48 | S | 74 | EI, myalgia, mild proximal | 5 | 98 | 1 | 109 | 0 | 0 | ||||||||||
| 36 | F/70 | D, G, S | n.d. | Mild proximal | 7 | 96 | 1 | 93 | n.d. | 3 | ||||||||||
| 43 | F/52 | D, G, H, L | 78 | Mild proximal | 6 | 96 | 1 | 133 | 1 | 2 | ||||||||||
| 46 | M/57 | C, G, HE | n.d. | Mild proximal, lower limbs | 6 | 97 | 2 | n.d. | n.d. | n.d. | ||||||||||
| Patients with ptosis | ||||||||||||||||||||
| 5 | F/44 | A, D, H, L, O, S | 67 | Ptosis, EI | 5 | 100 | 1 | 206 | 1 | n.d. | ||||||||||
| 8 | F/38 | C, G, H, S | 48 | Ptosis | 4 | 100 | 1 | 82 | 0 | n.d. | ||||||||||
| 10 | F/38 | G | 48 | Ptosis | 4 | 100 | 1 | 106 | 0 | n.d. | ||||||||||
| 24 | M/57 | D, G, H | 73 | Ptosis, EI, myalgia | 6 | 100 | 2 | 221 | 1 | 0 | ||||||||||
| Patient | Gender/age (years) | Clinical features | HP (%) | Myopathy characteristics | Decade of mp | MRC | Rankin score | CK (U/l) | RRF | CT | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patients with moderate limb weakness | ||||||||||||||||||||
| 2 | F/68 | G, H, L, S | 53 | Moderate proximal, mild waddling, myalgia | 5 | 82.5 | 2 | 177 | 1 | 2 | ||||||||||
| 7 | F/55 | G, HA, L, S | 67 | Moderate proximal, facial, asthenic | 5 | 82 | 2 | 144 | 1 | 3 | ||||||||||
| 11 | F/40 | B, C, E, G, H, L, N, R | 73 | Moderate proximal, waddling, asthenic | 3 | 85 | 3 | n.d. | 3 | n.d. | ||||||||||
| 13 | M/61 | G | 64 | Moderate proximal | 7 | 80 | 3 | 74 | 3 | n.d. | ||||||||||
| 15 | M/53 | A, D, G, L, N, S | 72 | Moderate general, facial, palatal, waddling | 4 | 82.5 | 3 | 358 | 0 | 3 | ||||||||||
| 16 | M/63 | C, D, G, H, L, N, S | 70 | Moderate proximal, ptosis | 5 | 89.5 | 3 | 188 | 3 | 2 | ||||||||||
| 18 | F/17 | E, G,HA, S | n.d. | Moderate general, facial | 2 | 81.5 | 2 | 57 | n.d. | 1 | ||||||||||
| 28 | F/65 | A, C, D, G, HE, L, N, S | 80 | Moderate general, facial, palatal, waddling, asthenic | 5 | 84 | 3 | 144 | 1 | n.d. | ||||||||||
| 29 | F/33 | D, E, G, HA, L, S | 86 | Moderate general, asthenic | 2 | 80 | 3 | 187 | 2 | n.d. | ||||||||||
| 30 | M/43 | C, S, N | 43 | Moderate proximal, facial, waddling, asthenic | 2 | 88.5 | 2 | n.d. | 0 | n.d. | ||||||||||
| 35 | M/65 | C, D, G, H, L, N, S | 57 | Moderate general, walking with assistance, asthenic | 5 | 89 | 4 | 122 | 1 | n.d. | ||||||||||
| 40 | F/62 | C, G, H, O, S | 66 | Moderate general, facial, waddling | 5 | 80.5 | 2 | 209 | 0 | 2 | ||||||||||
| 45 | M/54 | A, D, L, N, O, S | 74 | Moderate general, ptosis, facial, waddling | 5 | 82 | 3 | n.d. | 2 | n.d. | ||||||||||
| Patients with mild limb weakness | ||||||||||||||||||||
| 4 | F/48 | D, G, L, S, V | 75 | Mild, only thighs | 5 | 97.5 | 2 | 214 | 1 | 0 | ||||||||||
| 6 | F/20 | E, G, HA, L, R, S, SE | n.d. | Mild proximal | 2 | 96 | 2 | 235 | n.d. | 3 | ||||||||||
| 9 | M/16 | C, G, H, S | 76 | Mild proximal | 2 | 95 | 2 | 134 | 1 | 0 | ||||||||||
| 12 | M/49 | L | n.d. | Mild proximal | 5 | 95 | 1 | 225 | n.d. | 1 | ||||||||||
| 19 | F/48 | S | 74 | EI, myalgia, mild proximal | 5 | 98 | 1 | 109 | 0 | 0 | ||||||||||
| 36 | F/70 | D, G, S | n.d. | Mild proximal | 7 | 96 | 1 | 93 | n.d. | 3 | ||||||||||
| 43 | F/52 | D, G, H, L | 78 | Mild proximal | 6 | 96 | 1 | 133 | 1 | 2 | ||||||||||
| 46 | M/57 | C, G, HE | n.d. | Mild proximal, lower limbs | 6 | 97 | 2 | n.d. | n.d. | n.d. | ||||||||||
| Patients with ptosis | ||||||||||||||||||||
| 5 | F/44 | A, D, H, L, O, S | 67 | Ptosis, EI | 5 | 100 | 1 | 206 | 1 | n.d. | ||||||||||
| 8 | F/38 | C, G, H, S | 48 | Ptosis | 4 | 100 | 1 | 82 | 0 | n.d. | ||||||||||
| 10 | F/38 | G | 48 | Ptosis | 4 | 100 | 1 | 106 | 0 | n.d. | ||||||||||
| 24 | M/57 | D, G, H | 73 | Ptosis, EI, myalgia | 6 | 100 | 2 | 221 | 1 | 0 | ||||||||||
HP = mutation heteroplasmy in muscle; Decade of mp = decade of life, during which symptoms of myopathy first occurred; MRC = composite Medical Research Council score; Rankin score, 1 = no significant disability, 2 = slight disability, 3 = moderate disability, 4 = moderately severe disability; CK = creatine kinase; RRF = ragged red fibres (0 = none, 1 = few, 2 = moderate number, 3 = many); CT = muscle computed tomography (0 = normal, 1 = mild, 2 = moderate, 3 = severe changes); F = female; M = male; A = ataxia; B = basal ganglia calcifications; C = cognitive decline; D = diabetes mellitus; E = epilepsy; EI = exercise intolerance; G = short stature; H = cardiac hypertrophy; HA = occasional headache; HE = hepatopathy; L = lactic acidosis; N = peripheral neuropathy; O = ophthalmoplegia; R = pigment retinopathy; S = sensorineural hearing impairment; SE = stroke-like episodes; V = vitiligo; n.d. = not determined.
The age at onset of myopathy could be determined from the history in 16 cases, while the onset was defined as the age at clinical examination in nine cases. The highest incidence of myopathy was found in the fifth decade of life and the cumulative incidence by the age of 70 years was 0.77 for the patients (Fig. 1).
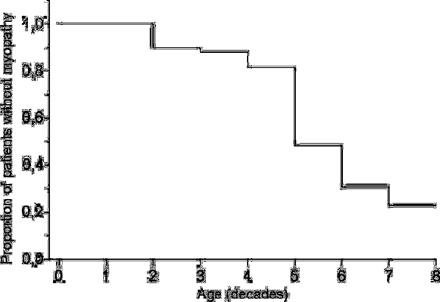
Proportion of patients remaining unaffected. Onset of myopathy was defined as the end-point. Unaffected patients were censored at the age of death or at their present age.
Nineteen patients (47%) reported active physical exercise, while 20% exercised occasionally and the remaining patients (33%) did not take any exercise. No difference in physical activity was found between the 13 myopathic patients with moderate limb weakness and the remaining patients. The patients with myopathy were significantly slower in standing up from the supine position than the non-myopathic ones, and their gait and stepping onto a footstool were frequently abnormal (Table 2). Myopathic patients with moderate limb weakness were slightly older than patients without limb weakness. There was a correlation between myopathy and neuropathy (P < 0.001) among the 50 patients, but not between myopathy and other clinical features such as short stature, ataxia, lactic acidosis, diabetes mellitus, cardiomyopathy, epilepsy or stroke-like episodes.
Comparison of the 3243A>G patients without any signs of myopathy with those having moderate limb weakness
| Patients without myopathy | Patients with moderate limb weakness | P value | |
|---|---|---|---|
| Men/women (n) | 8/17 | 6/7 | 0.50 |
| Age (years) | 32 | 55 | 0.03 |
| Diabetes (n) | 10 | 6 | 0.75 |
| Modified Rankin score | 1 (0–3) | 2 (2–3) | <0.001* |
| Composite MRC score | 100.0 | 82.5 | <0.001* |
| Muscle heteroplasmy (%) | 82 | 68 | 0.35 |
| Physical activity score | 2 (0–2) | 1 (0–2) | 0.14 |
| Standing up time (s) | 4 | 15 | <0.001* |
| Abnormal stepping test (n) | 4 | 13 | <0.001* |
| Gait abnormality (n) | 1 | 10 | <0.001* |
| Blood lactate (mmol/l) | 1.4 | 2.4 | 0.17 |
| Elevated serum CK (n) | 5 | 3 | 1.00 |
| Ragged red fibres (score) | 1 (0–3) | 1 (0–3) | 0.12 |
| COX-negative fibres (%) | 0.4 | 4.0 | 0.001* |
| Patients without myopathy | Patients with moderate limb weakness | P value | |
|---|---|---|---|
| Men/women (n) | 8/17 | 6/7 | 0.50 |
| Age (years) | 32 | 55 | 0.03 |
| Diabetes (n) | 10 | 6 | 0.75 |
| Modified Rankin score | 1 (0–3) | 2 (2–3) | <0.001* |
| Composite MRC score | 100.0 | 82.5 | <0.001* |
| Muscle heteroplasmy (%) | 82 | 68 | 0.35 |
| Physical activity score | 2 (0–2) | 1 (0–2) | 0.14 |
| Standing up time (s) | 4 | 15 | <0.001* |
| Abnormal stepping test (n) | 4 | 13 | <0.001* |
| Gait abnormality (n) | 1 | 10 | <0.001* |
| Blood lactate (mmol/l) | 1.4 | 2.4 | 0.17 |
| Elevated serum CK (n) | 5 | 3 | 1.00 |
| Ragged red fibres (score) | 1 (0–3) | 1 (0–3) | 0.12 |
| COX-negative fibres (%) | 0.4 | 4.0 | 0.001* |
Values are medians. MRC = Medical Research Council; Physical activity, 0 = no activity, 1 = occasionally, 2 = regularly; CK = creatine kinase; ragged red fibres, 0 = none, 1 = few, 2 = moderate number, 3 = many. The Mann–Whitney test was used. P values show the original values.
denotes significance.
Comparison of the 3243A>G patients without any signs of myopathy with those having moderate limb weakness
| Patients without myopathy | Patients with moderate limb weakness | P value | |
|---|---|---|---|
| Men/women (n) | 8/17 | 6/7 | 0.50 |
| Age (years) | 32 | 55 | 0.03 |
| Diabetes (n) | 10 | 6 | 0.75 |
| Modified Rankin score | 1 (0–3) | 2 (2–3) | <0.001* |
| Composite MRC score | 100.0 | 82.5 | <0.001* |
| Muscle heteroplasmy (%) | 82 | 68 | 0.35 |
| Physical activity score | 2 (0–2) | 1 (0–2) | 0.14 |
| Standing up time (s) | 4 | 15 | <0.001* |
| Abnormal stepping test (n) | 4 | 13 | <0.001* |
| Gait abnormality (n) | 1 | 10 | <0.001* |
| Blood lactate (mmol/l) | 1.4 | 2.4 | 0.17 |
| Elevated serum CK (n) | 5 | 3 | 1.00 |
| Ragged red fibres (score) | 1 (0–3) | 1 (0–3) | 0.12 |
| COX-negative fibres (%) | 0.4 | 4.0 | 0.001* |
| Patients without myopathy | Patients with moderate limb weakness | P value | |
|---|---|---|---|
| Men/women (n) | 8/17 | 6/7 | 0.50 |
| Age (years) | 32 | 55 | 0.03 |
| Diabetes (n) | 10 | 6 | 0.75 |
| Modified Rankin score | 1 (0–3) | 2 (2–3) | <0.001* |
| Composite MRC score | 100.0 | 82.5 | <0.001* |
| Muscle heteroplasmy (%) | 82 | 68 | 0.35 |
| Physical activity score | 2 (0–2) | 1 (0–2) | 0.14 |
| Standing up time (s) | 4 | 15 | <0.001* |
| Abnormal stepping test (n) | 4 | 13 | <0.001* |
| Gait abnormality (n) | 1 | 10 | <0.001* |
| Blood lactate (mmol/l) | 1.4 | 2.4 | 0.17 |
| Elevated serum CK (n) | 5 | 3 | 1.00 |
| Ragged red fibres (score) | 1 (0–3) | 1 (0–3) | 0.12 |
| COX-negative fibres (%) | 0.4 | 4.0 | 0.001* |
Values are medians. MRC = Medical Research Council; Physical activity, 0 = no activity, 1 = occasionally, 2 = regularly; CK = creatine kinase; ragged red fibres, 0 = none, 1 = few, 2 = moderate number, 3 = many. The Mann–Whitney test was used. P values show the original values.
denotes significance.
Laboratory data
Serum creatine kinase (CK) activity was mildly elevated in 14 of 44 (32%) examined patients (five men, nine women, range 171–654 U/l) and, interestingly, these patients often had lactic acidosis (P = 0.007), diabetes (P = 0.003) or CNS manifestations (P = 0.004). Venous blood lactate at rest was elevated in 47% of the patients and was higher among the men (mean 2.0 mmol/l, range 0.6–4.5) than among the women (mean 1.6 mmol/l, range 0.5–5.1). Increased blood lactate was associated with high mutation heteroplasmy (P = 0.002) and diabetes (P < 0.001), but we found no association between clinical myopathy and elevated lactate or CK activity.
Mutation heteroplasmy in muscle
Mutation heteroplasmy in the muscle varied from 30 to 94% (mean heteroplasmy 69 ± 14%), but no difference was found between the myopathic and non-myopathic patients or between those with mild and moderate myopathy. Heteroplasmy was higher among the patients with CNS manifestations [79%; 95% confidence interval (CI), 73–85%] than among those without (65%; 95% CI, 59–71%), and higher among the patients with diabetes (77%; 95% CI, 73–82%) than among those without (61%; 95% CI, 54–68%).
Histological and ultrastructural findings
Muscle histology was abnormal in 26 out of 36 patients (72%). Myopathic findings were seen in nine of these (25%), atrophic changes in eight (22%), fat infiltration in three (8%) and inflammatory changes in one (3%). RRFs were absent in 34% of the muscle samples, few in 43%, moderate in 9% and numerous in 14%. The presence of numerous RRFs did not correlate with the severity of myopathy, but it did correlate with features such as diabetes mellitus (P = 0.001), epilepsy (P = 0.006) and lactic acidosis (P = 0.001). The mean proportion of COX-negative fibres in the muscle of the 16 patients that were analysed was 1.8% (range 0–5.0), and a significant difference in this proportion was found between the patients without myopathy and those with myopathy regardless of its severity (Table 2).
Ultrastructural examination of muscle samples revealed that variations in mitochondrial size and shape were common (Fig. 2). Furthermore, both types of crystals were present frequently in these patients.
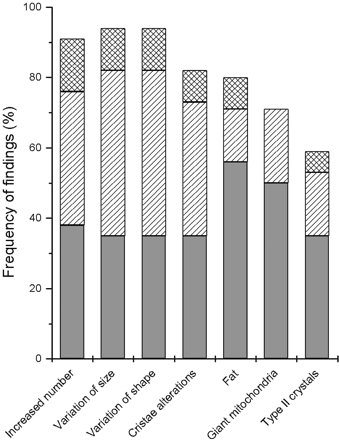
Frequency of ultrastructural findings in 34 patients with 3243A>G. Grey bars = proportion of mild changes; hatched bars = moderate changes; cross-hatched bars = severe changes.
Single fibre studies
The mean proportion of mtDNA with 3243A>G was 50% (range 3–92) in 251 histologically normal fibres from the 10 patients (Table 3). Eight of these patients had no muscle weakness, one had moderate weakness and one had only ptosis. The mean of mutation heteroplasmy was 86% (range 58–97) in COX-positive RRFs and slightly higher in the COX-negative ones (mean 90%, range 81–97).
Clinical and histological features of the patients with 3243A>G examined for single muscle fibres
| Patient | Age (years) | Gender | Clinical features | Frequency of fibres (%) | Mutation heteroplasmy (%) | ||
|---|---|---|---|---|---|---|---|
| RRF | COX-negative | ||||||
| 10 | 38 | F | G, P | 0 | 0.2 | 38.0 ± 3.1 | |
| 14 | 35 | F | 0 | 0 | 26.7 ± 2.6 | ||
| 21 | 39 | F | B, C, D, L, S | 0 | 1.2 | 32.0 ± 4.5 | |
| 22 | 33 | F | 0 | 0 | 50.4 ± 2.9 | ||
| 25 | 37 | F | H, S | 0 | 0.1 | 40.8 ± 4.7 | |
| 27 | 45 | M | C, S | 0.1 | 0.4 | 45.6 ± 5.0 | |
| 37 | 37 | M | C, G, S§ | 1.8 | 0.2 | 64.6 ± 2.4 | |
| 39 | 34 | F | B, G, H, L, R, S | 0.2 | 0.3 | 62.2 ± 2.7 | |
| 40 | 62 | F | C, O, G, H, M, S | 1.3 | 1.1 | 58.4 ± 2.9 | |
| 42 | 24 | M | C, D, E, G, L, S | 6.8 | 1.9 | 65.4 ± 3.0 | |
| Patient | Age (years) | Gender | Clinical features | Frequency of fibres (%) | Mutation heteroplasmy (%) | ||
|---|---|---|---|---|---|---|---|
| RRF | COX-negative | ||||||
| 10 | 38 | F | G, P | 0 | 0.2 | 38.0 ± 3.1 | |
| 14 | 35 | F | 0 | 0 | 26.7 ± 2.6 | ||
| 21 | 39 | F | B, C, D, L, S | 0 | 1.2 | 32.0 ± 4.5 | |
| 22 | 33 | F | 0 | 0 | 50.4 ± 2.9 | ||
| 25 | 37 | F | H, S | 0 | 0.1 | 40.8 ± 4.7 | |
| 27 | 45 | M | C, S | 0.1 | 0.4 | 45.6 ± 5.0 | |
| 37 | 37 | M | C, G, S§ | 1.8 | 0.2 | 64.6 ± 2.4 | |
| 39 | 34 | F | B, G, H, L, R, S | 0.2 | 0.3 | 62.2 ± 2.7 | |
| 40 | 62 | F | C, O, G, H, M, S | 1.3 | 1.1 | 58.4 ± 2.9 | |
| 42 | 24 | M | C, D, E, G, L, S | 6.8 | 1.9 | 65.4 ± 3.0 | |
F = female; M = male; RRF = ragged red fibre; COX = cytochrome c oxidase; B = basal ganglia calcifications; C = cognitive decline; D = diabetes mellitus; E = epilepsy; G = short stature; H = cardiac hypertrophy; L = lactic acidosis; M = clinical myopathy; O = ophthalmoplegia; P = ptosis; R = pigment retinopathy; S = sensorineural hearing impairment (§deaf). Mutation heteroplasmy is the proportion of 3243A>G in histologically normal muscle fibres (n = 37 for patient 42; n = 18–29 for other patients) (mean ± SEM). The men were unrelated, whereas the women were a family of a mother and her six daughters.
Clinical and histological features of the patients with 3243A>G examined for single muscle fibres
| Patient | Age (years) | Gender | Clinical features | Frequency of fibres (%) | Mutation heteroplasmy (%) | ||
|---|---|---|---|---|---|---|---|
| RRF | COX-negative | ||||||
| 10 | 38 | F | G, P | 0 | 0.2 | 38.0 ± 3.1 | |
| 14 | 35 | F | 0 | 0 | 26.7 ± 2.6 | ||
| 21 | 39 | F | B, C, D, L, S | 0 | 1.2 | 32.0 ± 4.5 | |
| 22 | 33 | F | 0 | 0 | 50.4 ± 2.9 | ||
| 25 | 37 | F | H, S | 0 | 0.1 | 40.8 ± 4.7 | |
| 27 | 45 | M | C, S | 0.1 | 0.4 | 45.6 ± 5.0 | |
| 37 | 37 | M | C, G, S§ | 1.8 | 0.2 | 64.6 ± 2.4 | |
| 39 | 34 | F | B, G, H, L, R, S | 0.2 | 0.3 | 62.2 ± 2.7 | |
| 40 | 62 | F | C, O, G, H, M, S | 1.3 | 1.1 | 58.4 ± 2.9 | |
| 42 | 24 | M | C, D, E, G, L, S | 6.8 | 1.9 | 65.4 ± 3.0 | |
| Patient | Age (years) | Gender | Clinical features | Frequency of fibres (%) | Mutation heteroplasmy (%) | ||
|---|---|---|---|---|---|---|---|
| RRF | COX-negative | ||||||
| 10 | 38 | F | G, P | 0 | 0.2 | 38.0 ± 3.1 | |
| 14 | 35 | F | 0 | 0 | 26.7 ± 2.6 | ||
| 21 | 39 | F | B, C, D, L, S | 0 | 1.2 | 32.0 ± 4.5 | |
| 22 | 33 | F | 0 | 0 | 50.4 ± 2.9 | ||
| 25 | 37 | F | H, S | 0 | 0.1 | 40.8 ± 4.7 | |
| 27 | 45 | M | C, S | 0.1 | 0.4 | 45.6 ± 5.0 | |
| 37 | 37 | M | C, G, S§ | 1.8 | 0.2 | 64.6 ± 2.4 | |
| 39 | 34 | F | B, G, H, L, R, S | 0.2 | 0.3 | 62.2 ± 2.7 | |
| 40 | 62 | F | C, O, G, H, M, S | 1.3 | 1.1 | 58.4 ± 2.9 | |
| 42 | 24 | M | C, D, E, G, L, S | 6.8 | 1.9 | 65.4 ± 3.0 | |
F = female; M = male; RRF = ragged red fibre; COX = cytochrome c oxidase; B = basal ganglia calcifications; C = cognitive decline; D = diabetes mellitus; E = epilepsy; G = short stature; H = cardiac hypertrophy; L = lactic acidosis; M = clinical myopathy; O = ophthalmoplegia; P = ptosis; R = pigment retinopathy; S = sensorineural hearing impairment (§deaf). Mutation heteroplasmy is the proportion of 3243A>G in histologically normal muscle fibres (n = 37 for patient 42; n = 18–29 for other patients) (mean ± SEM). The men were unrelated, whereas the women were a family of a mother and her six daughters.
Mutation heteroplasmy was then determined in 10 muscle sections spanning a longitudinal length of 200 µm in patient 42 (Fig. 3A and B) and patient 37 (Fig. 3C and D). The sections harboured two COX-positive RRFs in each patient and, on average, five histologically normal fibres adjacent to the RRFs. The total variation in mutation heteroplasmy along the four RRFs was small, but there were five 10 µm segments, where the mutation heteroplasmy was clearly lower (median 68%, range 64–74) than that in the neighbouring segments. Furthermore, histologically normal fibres showed segmental distribution of the 3243A>G mutation in patient 42. Normal fibres surrounding RRF 1 in sections 1, 7 and 8 (Fig. 3A) harboured heteroplasmy above the average (P = 0.08 for the difference between sections, Kruskal–Wallis test), while normal fibres surrounding RRF 2 in section 6 (Fig. 3B) harboured heteroplasmy below the average (P = 0.06 for the difference between sections). The variations in patient 37 were not remarkable (Fig. 3C and D). Interestingly, among the sections, a correlation was found between mutation heteroplasmy in the RRF and that in the adjacent histologically normal fibres. In sections harbouring RRF with a mutation heteroplasmy ≤86%, the adjacent normal fibres had a lower proportion of the mutation than normal fibres in sections harbouring RRF with a mutation heteroplasmy >86% (P = 0.05).
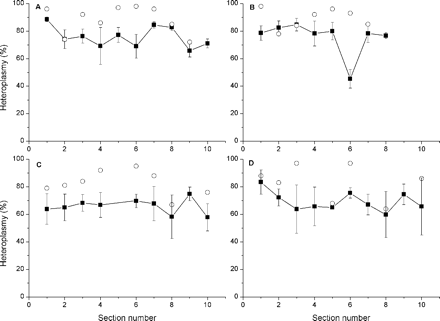
Mutation heteroplasmy in ragged red fibres and adjacent histologically normal fibres in 10 consecutive cross-sections of muscle. The mean proportion (±SEM) of the 3243A>G mutation is shown. A = RRF 1 from patient 42; B = RRF 2 from patient 42; C = RRF 1 from patient 37; D = RRF 2 from patient 37. Solid symbols = histologically normal fibres adjacent to RRF; open symbols = ragged red fibres.
Discussion
We found clinically defined myopathy in 25 out of 50 patients identified in a population-based screening as possessing the 3243A>G mutation. Most of the patients had mild or moderate limb weakness, while others had ptosis and/or other muscle symptoms. The patients with moderate limb weakness considered myopathy a major symptom. In the cases of limb involvement, the characteristic clinical features consisted of symmetric proximal myopathy, and a waddling gait was observed in most of these patients as a result of weakness in the hip abductors. Involvement of the pelvic muscles in fact is a common finding in CT of muscles among 3243A>G patients (Kärppä et al., 2004) and EMG abnormalities are more frequent in proximal muscles, although they occur less abundantly than changes in CT scans (Kärppä et al., 2004). Limb weakness, usually mild, has been suggested as being one of the most common clinical manifestations among these patients (Hammans et al., 1995). In addition, we found that ptosis or ophthalmoplegia were not unusual, although it has been considered to be atypical of patients with 3243A>G (Moraes et al., 1993). The myopathic patients obtained lower grades in the global functional assessment, but cardiomyopathy, stroke-like episodes or other severe features were no more common among them than among the non-myopathic patients. These findings suggest that their functional disability was essentially a result of the myopathy itself.
Our observation was that the risk of myopathy increased with age. A higher age has been shown to be associated with an increased risk of peripheral neuropathy (Kärppä et al., 2003) and increased severity of hearing impairment in patients with 3243A>G (Uimonen et al., 2001). We found that the highest incidence of myopathy in patients with 3243A>G was in the fifth decade of life, suggesting that the decline in oxidative phosphorylation in the skeletal muscle had reached the threshold of clinical manifestation. Skeletal muscle undergoes age-related changes such as a shift in the proportion of fibre types towards type I, neurogenic muscular atrophy and other structural and histochemical alterations, and, furthermore, a clear decline has been reported in respiratory chain function in normal muscle (Brierley et al., 1997). It has been suggested that a progressive accumulation of COX-deficient muscle fibres may be part of the natural history of mitochondrial myopathy (Weber et al., 1997) and a random accumulation of COX-negative muscle fibres has been documented in association with the clinical progression of mitochondrial myopathy (Chinnery et al., 2003). Ageing in itself does not appear to impair mitochondrial function in healthy elderly athletes, however (Brierley et al., 1996), and it has been suggested that altered physical activity may also contribute to the decline (Boffoli et al., 1994). We found that physical inactivity is not associated with myopathy.
No correlation was found between mutation heteroplasmy and the presence of myopathy. In fact, the myopathic patients showed slightly lower mean heteroplasmy, while a higher heteroplasmy was associated with CNS manifestations and diabetes. The patients with a higher muscle 3243A>G mutation load may be affected with encephalopathy at a young age, whereas those with a lower mutation load present with myopathy in later years. The finding of a peak incidence of myopathy in the fifth decade would support this assumption. Furthermore, an inverse correlation between mutation load and muscle symptoms has been observed in a meta-analysis based on 117 published cases (Chinnery et al., 1997).
COX-negative fibres and histologically normal fibres make a mosaic pattern in cross-sections of muscle from patients with mitochondrial myopathies (Brierley et al., 1997; Chinnery et al., 1997; Larsson and Oldfors, 2001). Longitudinally, COX-negative segments have been found to exceed 1 mm in length (Matsuoka et al., 1991; Elson et al., 2002) and, interestingly, segments of 10 µm with intermediate or normal COX activity have been found to interrupt long stretches of COX-negative fibres in a patient with the common mtDNA deletion (Elson et al., 2002). Direct comparison of histochemical dysfunction in COX-negative fibres from a patient with the common mtDNA deletion and mutation load in COX-positive RRFs from patients with 3243A>G may be biased. However, it was interesting to find evidence for longitudinal variations in heteroplasmy in COX-positive RRFs and in histologically normal muscle fibres of patients with the common MELAS mutation. The 3243A>G mutation heteroplasmy was high and fairly constant in four RRFs, but within a total length of 800 µm we found five 10 µm segments where the heteroplasmy was clearly lower than that in the neighbouring sections.
We observed evidence for variation between longitudinal sections in histologically normal muscle fibres as well. Most interestingly, the load of 3243A>G in a 10 µm segment in patient 42 differed from the mean load of mutation along the fibre. Most of the histologically normal fibres harboured a mutation load that was clearly below the threshold of biochemical dysfunction, suggesting that selection may not play an important role. Interestingly, the mutation load in histologically normal fibres adjacent to an RRF seemed to depend on that in the adjacent RRF itself. Mitotic segregation during organogenesis may contribute to this finding, but perinuclear clustering of mitochondria has been shown to take place in cultured cells following the inhibition of the molecular motor kinesin, which is driven by ATP (Ebneth et al., 1998). Segmental reduction in cellular energy could lead to clustering of the organelles, suggesting a basis for the non-random distribution of mutant and wild-type mtDNAs which causes segmental defects in all kinds of muscle fibres, i.e. in normal-looking fibres, COX-negative fibres and RRFs.
We detected significantly more COX-negative muscle fibres and intramitochondrial crystals in the limb weakness patients than in those without limb weakness. The more COX-negative fibres were present in muscle histochemistry, the more severe was the myopathy clinically, whereas the number of RRFs did not predict the severity of myopathy, even though an increased number of mitochondria and increased variation in their size and shape were found in the muscle of >90% of the patients. This finding is probably due to the fact that the enzyme defect reflects the myopathic process more accurately than does mitochondrial proliferation. An increased number of mitochondria is usually a sign of a mitochondrial disease, and large subsarcolemmal aggregates of mitochondria are the most constant ultrastructural finding in mitochondrial diseases (Morgan-Hughes, 1994).
We found that 32% of patients had elevated CK. Myofibre necrosis or membrane leakage are the major causes of serum CK elevation, and increased serum CK levels usually correlate well with the extent and severity of myopathy. Increased CK has been reported to correlate with limb weakness in patients with 3243A>G (Sciacco et al., 2001). We found that CK was mildly elevated only in one-third of the patients and that their CK values did not correlate with the severity of myopathy, indicating that fibre necrosis or increased sarcolemmal permeability are not common in patients with 3243A>G. We found associations between increased CK and diabetes, lactic acidosis and CNS manifestations. The significance of these findings remains unclear.
We are reporting here on the largest clinical study so far on the occurrence of myopathy among patients with the 3243A>G mtDNA mutation. The patients had been identified in a defined population, and they therefore represent a good approximation to a population-based cohort. We found that a clinical diagnosis of myopathy could be made in 50% of the patients, whereas the histological muscle examination was abnormal in 72% of the cases. Moderate limb weakness was the most common myopathic symptom, but mild weakness and external ophthalmoplegia could also be found. The highest incidence of myopathy was in the fifth decade of life, but no other clinical variables apart from age were associated with an increased risk of myopathy. Histological and ultrastructural abnormalities were present both in the patients with myopathy and in those without myopathy, but the presence of crystals and COX-negative fibres and variation in the size and shape of the mitochondria were more common in the muscle of myopathic patients. We conclude that myopathy is highly variable in presentation in patients with 3243A>G.
We wish to thank Ms Pirjo Keränen for her expert technical assistance. This work was supported in part by grants from the Sigrid Juselius Foundation, Oy Lundbeck Ab and the Medical Research Council of the Academy of Finland.
References
Boffoli D, Scacco SC, Vergari R, Solarino G, Santacroce G, Papa S. Decline with age of the respiratory chain activity in human skeletal muscle.
Brierley E, Johnson M, James O, Turnbull D. Effects of physical activity and age on mitochondrial function.
Brierley EJ, Johnson MA, Bowman A, Ford GA, Subhan F, Reed JW, et al. Mitochondrial function in muscle from elderly athletes.
Chinnery PF, Howell N, Lightowlers RN, Turnbull DM. Molecular pathology of MELAS and MERFF. The relationship between mutation load and clinical phenotypes.
Chinnery PF, Howel D, Turnbull DM, Johnson MA. Clinical progression of mitochondrial myopathy is associated with the random accumulation of cytochrome c oxidase negative skeletal muscle fibers.
Cwik VA, Brooke MH. Proximal, distal and generalized weakness. In: Bradley WG, Daroff RB, Fenichel GM, Marsden CD, editors. Neurology in clinical practice. Butterworth-Heinemann;
Deschauer M, Müller T, Wieser T, Schulte-Mattler W, Kornhuber M, Zierz S. Hearing impairment is common in various phenotypes of the mitochondrial DNA A3243G mutation.
Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E, et al. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease.
Elson JL, Samuels DC, Johnson MA, Turnbull DM, Chinnery PF. The length of cytochrome c oxidase negative segments in human skeletal muscle fibres of patients with a mitochondrial myopathy.
Goto Y, Nonaka I, Horai S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies.
Hammans SR, Sweeney MG, Hanna MG, Brocklington M, Morgan-Hughes JA, Harding AE. The mitochondrial DNA transfer RNALeu(UUR) A>G(3243) mutation. A clinical and genetic study.
Katirji B. Clinical assessment in neuromuscular disorders. In: Katirji B, Kaminski HJ, Preston DC, Ruff RL, Shapiro BE, editors. Neuromuscular disorders in clinical practice. Butterworth-Heinemann;
Kobayashi Y, Momoi MY, Tominaga K, Momoi, Nihei K, Yanagisawa M, et al. A point mutation in the mitochondrial tRNA(Leu)(UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes).
Kärppä M, Syrjälä P, Tolonen U, Majamaa K. Peripheral neuropathy in patients with the 3243A>G mutation in mitochondrial DNA.
Kärppä M, Mahjneh I, Karttunen A, Tolonen U, Majamaa K. Muscle computed tomography patterns in patients with the mitochondrial DNA mutation 3243A>G.
Majamaa K, Moilanen JS, Uimonen S, Remes AM, Salmela PI, Kärppä M, et al. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: prevalence of the mutation in an adult population.
Matsuoka T, Goto Y, Yoneda M, Nonaka I. Muscle histopathology in myoclonus epilepsy with ragged-red fibers (MERRF).
Moraes CT, Ciacci F, Silvestri G, Shanske S, Sciacco M, Hirano M, et al. Atypical clinical presentations associated with the MELAS mutation at position 3243 of human mitochondrial DNA.
Morgan-Hughes JA. Mitochondrial diseases. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw-Hill;
Moslemi AR, Tulinius M, Holme E, Oldfors A. Threshold expression of the tRNA(Lys) A8344G mutation in single muscle fibres.
Oldfors A, Moslemi AR, Fyhr IM, Holme E, Larsson NG, Lindberg C. Mitochondrial DNA deletions in muscle fibers in inclusion body myositis.
Petruzzella V, Moraes CT, Sano MC, Bonilla E, DiMauro S, Schon EA. Extremely high levels of mutant mtDNAs co-localize with cytochrome c oxidase-negative ragged-red fibers in patients harboring a point mutation at nt 3243.
Sciacco M, Bonilla E, Schon EA, DiMauro S, Moraes CT. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy.
Sciacco M, Prelle A, Comi GP, Napoli L, Battistel A, Bresolin N, et al. Retrospective study of a large population of patients affected with mitochondrial disorders: clinical, morphological and molecular genetic evaluation.
Shoffner JM, Lott MT, Lezza AM, Seibel P, Ballinger SW, Wallace DC. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation.
Stadhouders AM, Jap PHK, Winkler HP, Eppenberger HM, Wallimann T. Mitochondrial creatine kinase: a major constituent of pathological inclusions seen in mitochondrial myopathies.
Uimonen S, Moilanen JS, Sorri M, Hassinen IE, Majamaa K. Hearing impairment in patients with 3243A>G mtDNA mutation: phenotype and rate of progression.
Author notes
1Department of Neurology, University of Oulu, 2Clinical Research Center, 3Department of Pathology, 4Department of Internal Medicine, Oulu University Hospital, Oulu, 5Department of Pathology, Sahlgrenska University Hospital, Gothenburg, Sweden and 6Department of Neurology, University of Turku, Turku, Finland


{kind=link}
{kind=link}
{kind=link}