
Abstract
We have analysed the splicing pattern of the human Duchenne Muscular Dystrophy (DMD) transcript in normal skeletal muscle. To achieve depth of coverage required for the analysis of this lowly expressed gene in muscle, we designed a targeted RNA-Seq procedure that combines amplification of the full-length 11.3 kb DMD cDNA sequence and 454 sequencing technology. A high and uniform coverage of the cDNA sequence was obtained that allowed to draw up a reliable inventory of the physiological alternative splicing events in the muscular DMD transcript. In contrast to previous assumptions, we evidenced that most of the 79 DMD exons are constitutively spliced in skeletal muscle. Only a limited number of 12 alternative splicing events were identified, all present at a very low level. These include previously known exon skipping events but also newly described pseudoexon inclusions and alternative 3′ splice sites, of which one is the first functional NAGNAG splice site reported in the DMD gene. This study provides the first RNA-Seq-based reference of DMD splicing pattern in skeletal muscle and reports on an experimental procedure well suited to detect condition-specific differences in this low abundance transcript that may prove useful for diagnostic, research or RNA-based therapeutic applications.
Similar content being viewed by others


Introduction
Alternative splicing (AS) is a key mechanism for generating tissue or developmental stage-specific proteomic diversity in eukaryotes1,2. Muscle was one of the first tissues in which AS was widely observed in particular in contractile protein genes3. Recent global analyses of splicing programs have confirmed that skeletal muscle is among the tissues showing the largest number of tissue-specific AS events (ASEs)4.
The DMD gene encodes dystrophin, a cytoskeletal protein of 427 kDa accounting for only approximately 0.002% of the total striated muscle protein content but playing an essential role in muscle fiber integrity and function. Loss-of-function mutations cause Duchenne muscular dystrophy (DMD), the most common and severe form of progressive muscular dystrophy in children5. The DMD transcript can be categorized among the low abundance transcripts relative to other muscle transcripts. In adult skeletal muscle tissues, its concentration is estimated to be 5–10 molecules per nucleus as compared to 25,000–50,000 copies per nucleus for the highly abundant muscle transcript encoding the myosin heavy chain6. The DMD gene is remarkable by its length (2.2 Mb, the longest human gene) and genomic structure. About 99% of the gene is made of introns, some of them being very long introns exceeding 200 kb, while the translated coding sequence, which is fragmented into 79 exons, is only 11.3 kb. Seven independent tissue-specific promoters encode three full-length isoforms (including the Dp427m muscle one) and four N-terminally truncated proteins5.
There is no clear picture of the global splicing profile of the muscle transcript. Available data are not readily comparable because techniques of different sensitivity were used in previous studies that generally focused only on specific regions of the gene7,8,9,10. Considering the currently developed therapeutic approaches for DMD based on antisense oligonucleotide-mediated splicing modulation11, as well as the importance of splicing defects as a cause or a modifier of disease severity12,13, there is a clear need to elucidate the full splicing pattern of the DMD transcript.
Massively parallel RNA sequencing (RNA-Seq) has become a powerful technology to explore the complexity of mammalian transcriptomes14. Besides allowing for the comparison of gene expression changes in response to cellular differentiation, environmental factors or disease conditions, RNA-Seq can be used to accurately identify novel isoforms, assess relative transcript abundances and detect alternative exon and splice site usage in tissues or cells15. However, this approach has not yet provided information for all genes uniformly. Indeed a large fraction of sequence reads in RNA-Seq experiments are used up by highly expressed transcripts, thereby lowering the ability to detect other transcripts present at low levels16. This limitation is particularly detrimental to splicing analysis, which requires basically more input data than for gene expression analysis since a read must include the AS region to count towards splicing analysis17. Thus detection of alternative splicing within low abundance genes remains challenging. In this study, we designed a suitable strategy that relies on a DMD-targeted RNA-Seq protocol and the 454 technology to allow accurate and comprehensive inventory of physiological ASEs of the DMD transcript in human skeletal muscle.
Results
Design of an optimized RNA-Seq protocol for human DMD transcript analysis
Before designing our experimental workflow, a preliminary analysis of RNA sequencing data of human skeletal muscle tissue available in the Illumina’s Body Map (IBM) transcriptome project18 had shown us that the read depth along the DMD transcript was globally insufficient (50X–200X) despite the huge amount of reads produced, to allow reliable detection of ASEs present at low level. This observation prompted us to set up a DMD-targeted RNA-Seq approach (Fig. 1) that combines the amplification of the full DMD coding sequence (11.3 kb) with the Roche 454 sequencing technology. Typically 454 reads are >300 bp long and are likely to span more than two exon junctions that helps to identify non-canonical splicing events. To assess the performance of our procedure, a first comparison has been made between the data obtained from the analysis of one skeletal muscle sample using our targeted approach (average read length: 387 bp, 15.8 Mb) and the data from the IBM project (human skeletal muscle RNA GSM759515, 2 × 50 bp paired end, 8.2 Gb). Visualization of the aligned reads to the DMD Dp427m isoform showed differences in DMD mRNA sequencing depth and coverage (Fig. 2). With the total mRNA sequencing technology, the mean depth per base was 142 (+/−82 SD) reads and a marked decrease of coverage at the 5′end of the transcript was noticed (about 1 kb is covered by less than 50 reads) due to a bias toward the identification of sequences from the 3′ end of DMD mRNA as previously described19. By using our DMD-targeted approach, a relatively uniform coverage was obtained with a mean depth of 1,363 (+/−302 SD) reads per base that would theoretically allow ASEs as low as 1% to be reliably detected (>10 reads).

Schematic depiction of the four-stage protocol developed for the analysis of the DMD gene splicing pattern by RNA-Seq including the sample preparation, sequencing, data analysis and biological validation. (RT) reverse transcription, (LR-PCR) Long Range-PCR, (QC) quality control, (IGV) Integrative Genome Viewer, (STAR) Spliced Transcripts Alignment to a Reference software, (PSI) Percent-Spliced-In, (ASEs) Alternative Splicing Events.

Reads from each library were mapped to the DMD transcript (Dp427m mRNA, NM_004006.2) using Bowtie2 read aligner and DMD mRNA coverage was visualized through the Integrative Genome Viewer (IGV). Shown are the IGV coverage plots from (dark grey) publicly available total mRNA-Seq data (Illumina Human Body Map project 2.0, skeletal muscle tissue, GEO sample GSM759515, 2 × 50 bp paired end, 8.2 G bases), and (light grey) DMD mRNA-Seq data generated in this study (skeletal muscle sample n°20316, Myobank).
The Spliced Transcripts Alignment to a Reference software (STAR), specially designed for aligning long reads20, was then used to identify DMD canonical splice junctions as well as potential new ones from junction-spanning reads. Compared to TopHat2, a popular mapper suitable for processing relatively short reads, STAR performed better both in the total mRNA-Seq and our DMD-targeted mRNA-Seq datasets (Table 1) for the positioning of the 78 canonical junctions and the discovery of alternative splice junctions as illustrated with pseudoexon 1a, an already known case of pseudoexon insertion in the DMD transcript21.
Splicing pattern of DMD transcript in skeletal muscle tissue
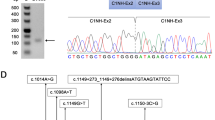
Four independent skeletal muscle tissue samples from normal young men were used to establish the splicing pattern of the muscular DMD transcript. We applied our established DMD-targeted RNA-Seq protocol, and the splice junction reads obtained both for the 78 canonical junctions and novel splice junctions in the 4 samples are detailed in the Supplementary Table S1. Among the non-canonical splice junctions detected, we chose to consider only those consistently detected with a minimum of 5 read counts in at least 2 out of the 4 muscle samples analysed for further analyses. Strikingly only 12 ASEs could be identified (Table 2), which can be divided into three different categories: exon skipping (n = 5), cryptic exon inclusion (n = 3) and 3′ alternative splice site (ss) usage (n = 4). Most of them (9/12) are low level ASEs (≤1%) and the three most represented events (del71, del78, PE1a) do not exceed 3%. It is noteworthy that the majority of them (66%, 8/12) preserve an open reading frame. While some events have already been reported (del9, del71, del78, PE1a), the other ones are here described for the first time (Supplementary Table S2). The very low level of detected ASEs precluded their experimental validation by RT-PCR in most cases, but we were able to conduct these analyses for three of them (del71, del78, and PE1a). We performed either standard or fluorescent RT-PCR using primers that amplify both the included and the skipped isoforms, and analysed PCR products by gel electrophoresis or fragment analysis on a capillary sequencer, respectively. The three ASEs were successfully validated (Fig. 3), with a greater concordance between RNA-Seq and fragment analysis data (Table 3).

Examples of RT-PCR validations of the three most represented ASEs by (a) standard RT-PCR and agarose gel electrophoresis or (b) capillary electrophoresis analysis of fluorescent PCR products. M, molecular-weight size marker. Quantification data (mean +/−SD) from at least three independent experiments are reported in Table 3. (c) Splice-site strength of pseudoexons. MaxEntScan (MES) and Human Splicing Finder (HSF) scores are given for alternative 3′ and 5′ splice sites (alt 3′ss, alt 5′ss) of pseudoexons 1a, 21X and 51X and for splice sites of adjacent constitutive exons (3′ss, 5′ss). The size of the intronic regions (in nucleotides, nt) flanking the pseudoexon is indicated in italics. (d) Splice-site strength of alternative 3′ splice sites. MES and HSF scores are given for exonic alternative 3′ splice sites (alt 3′ss) and adjacent authentic 3′ splice sites (3′ss). nd, not detected. The vertical dotted lines mark the position of the alternative 3′ss in exons 20, 54, 76 and 78. The number of deleted nucleotides (nt, in italics) in the mature dystrophin transcript when the alternative 3′ss is used is indicated on top of each exon.
In addition to exon skipping events, the RNA-Seq analysis has revealed the usage of alternative splice sites located either at proximity of the natural ones or deep in the introns (Table 2). The reliability of the bioinformatics analysis for these new sites was systematically verified by inspection of the raw sequencing reads. In particular for pseudoexons, we verified that the whole sequence of the pseudoexon could be retrieved from a single read. Erroneously annotated alternative splice sites were found at the exons 3/4 and exons 14/15 junctions due to the presence of homopolymers and/or sequence homologies that led to misalignment (Supplementary Table S1). These alternative junctions were thus excluded from our analysis. With the exception of the PE1a, the 6 other reported events (2 pseudoexons, and 4 alternative 3′ ss) are newly described in this study (Table 2). The strenght of these alternative splice sites was evaluated by maximum entropy scores (MaxEntScan, MES) and the Human Splicing Finder (HSF) program (Fig. 3).
All three pseudoexons displayed high 5′ and 3′ splice site scores, thus supporting their use to some extent in the DMD transcripts. Regarding the detected 3′ alternative ss, all are exonic and their calculated scores were lower than the scores of the adjacent natural splices in all cases except for the exon 76 + 59 alternative 3′ss, which exhibits scores (HSF: 89.39 and MES: 9.59) significantly higher than the natural 3′ss of exon 76 (HSF: 69.15 and MES: 3.81) (Fig. 3). When used, a new transcript is produced carrying an in-frame deletion of 20 amino acids (Pro3600 to Gln3619) in the C-terminal domain of dystrophin that is not predicted to alter a protein-protein interaction domain. The RNA-Seq analysis also disclosed splicing at a previously undescribed NAGNAG tandem site in DMD exon 54 that deletes one amino acid (p.Glu2625). This novel junction was reliably detected in all four analysed skeletal muscle samples (11–28 junction reads).
Lastly, we have drawn up a list of all detected exon skipping events but that failed to reach the minimum criteria set (i.e. a minimum of 5 reads in 2 out of the 4 samples analysed) (Supplementary Table S1). Interestingly we noticed that half of them (13/25, 52%) are located in the contiguous block of the 20 in-frame exons, encoding portions of the central rod domain of dystrophin, and the most represented ones (del25, del28, del28 + 29, del38) match with exons frequently involved in nonsense-induced exon skipping events that contribute to modify disease severity in patients. This observation further supports the idea that these exons have peculiar features regarding splicing, which may make them much more prone to exon skipping when mutated in patients22.
Discussion
Over half of the multi-exonic human genes are believed to have splice variants23. Due to its large number of exons, the DMD gene was a priori a good candidate for the occurrence of multiple alternative splicing events. A literature survey supports this common assumption with many reports describing alternative splicing events in the DMD gene since its discovery in 1986 (Supplementary Table S2). However these data derive from studies that are very heterogeneous in terms of the techniques used (northern blot, PCR, nested PCR), specificity of the transcript isoform analysed (Dp427m and/or other isoforms), biological samples analysed (human versus animal tissues, skeletal muscle versus other tissues or lymphocytes, patients versus normal controls), and gene regions explored (most studies focused on specific regions), that makes qualitative and quantitative comparisons between these different datasets difficult. A comprehensive and unbiased inventory of the DMD splicing events in skeletal muscle was lacking.
The recently developed deep-sequencing technologies allows a far more precise quantification of transcript levels and their isoforms than other methods14. In this study, a targeted RNA-Seq procedure was chosen to increase the sequencing depth and therefore to allow reliable detection of alternative splice junctions in the DMD gene. The deep RNA sequencing strategy was reasonably expected to expand the repertoire of ASEs in the DMD gene. Yet surprisingly, the vast majority of the 79 DMD exons were found to be constitutively spliced in skeletal muscle under physiological conditions. Hence, our results would indicate that the frequency of ASEs previously reported in the DMD muscular transcript may have been overestimated, possibly due to methodological considerations.
Exon-skipping was the most common form of the ASEs detected (5/12, 41.7%). Notwithstanding the large number of exons in the DMD gene, skipping of only a single or a limited number of exons were characterized (Table 2 and Supplemental Table 1). Skipping of long stretches of exons were not found in normal human skeletal muscle contrary to what can be observed in pathological condition, where multiple exon skipping rearrangements have been reported to occur, notably in revertant fibers to restore dystrophin expression in patients24 or in the mdx model25. Though few in number, the identity of the identified ASEs in this study provides strong support for the reliability of our RNA-Seq analysis. Indeed, several of them correspond to previously described ASEs occurring in a tissue-specific or developmental stage-specific manner in the full-length muscular transcript or in other isoforms9,26,27. In particular, exon 71 and exon 78 are alternatively spliced in Dp71, one of the shortest dystrophin isoform and the major DMD gene product in many non-muscle tissues. DMD transcripts alternatively spliced at the 3′ end encode functionally distinct protein isoforms that are likely to remodel protein-protein interaction networks28 in particular with components of the dystrophin-glycoprotein complex (DGC). The in-frame skipping of exon 71 occurs in close proximity to the gene region (exons 73–75) encoding the syntrophin-binding sites29, while the absence of exon 78 causes a frameshiſt that replaces the 13 C-terminal dystrophin amino acids residues with 31 new ones defining a protein with a novel hydrophobic carboxy terminus30. Exon 78 is spliced out in the embryonic muscle dystrophin isoform. This developmentally regulated alternative splicing is highly conserved throughout vertebrates, arguing for a critical functional role of this longer C-terminal protein domain during the embryonic stage of development. By contrast, dystrophin exon 78 is required for muscle structure maintenance in adult skeletal muscle. Its abnormal exclusion in patients suffering from myotonic dystrophy (DM) due to depletion of the MBNL1 splicing factor likely contributes to the progressive dystrophic process in DM type1 patients31. In accordance with these data, exon 78 was found to be included in about 98% of the muscular transcripts in our RNA-Seq experiments as was exon 71. The traces of exon skipping detected in muscle may reflect the complexity of the mechanisms controlling the inclusion of these two exons in a tissue-specific or developmental stage-specific manner9,32. It is noteworthy that DMD exon 71 and exon 78 are very short exons (39 and 32 bp, respectively) that share some characteristics with the recently described microexons in neurons, a set of highly conserved short exons, which are strongly regulated by RNA-binding proteins (RBPs) and functionally modulate tissue-dependent protein-protein networks33,34.
The RNA-Seq analysis has revealed the usage of alternative splice sites located either in the close vicinity of the natural sites or deep in the introns (Table 2), which are all newly described except pseudoexon 1a. The pseudoexon 1a originating from intron 1 was initially identified in lymphocytes21, where it is included in more than 50% of the illegitimate DMD transcripts. The inclusion of this out-of-frame extra-exon may represent a post-transcriptional control in cells that normally do not express the dystrophin protein (like lymphocytes), while a low inclusion rate is observed in skeletal muscle transcripts (3%). Due to the size of the DMD introns, multistep splicing events (recursive or nested splicing) are frequently used to splice out a single intron35. Interestingly, the 5′ and 3′ genomic coordinates of the identified pseudoexons in our study (1a, 21X, 51X) are similar to 6 out of the 145 biocomputational predicted positions associated with either 5′ or 3′ recursive splicing of multi-step intron removal recently identified by capture-pre-mRNA-seq of intermediately spliced dystrophin transcript35. Whether these 6 positions represent true motifs required for 5′and 3′ recursive splicing remains to be experimentally demonstrated, but our findings show that they can be used in combination for the inclusion of pseudoexons in the mature transcripts.
As previously reported in other human genes36, we observed that alternative acceptors were the second most common (4/12, 33%) type of AS detected in DMD after exon skipping. All are exonic alternative 3′ splice sites that induce partial exon deletion when used, and the major one involves a NAGNAG tandem site in DMD exon 54. NAGNAG motif occurs in 30% of human genes and appears to be functional in at least 5% of human genes37,38. Their use results in the production of the two distinct isoforms distinguishable by three nucleotides (NAG). These subtle changes may nonetheless be of functional relevance by changing local hydrophobicity and charge, varying the distances between relevant sites in proteins or changing recognition sequences for post-translational modifications. This is the first functional NAGNAG motif identified in the DMD gene. Splicing at this site deletes the polar amino acid glutamine at position 2625 of dystrophin repeat 21 which forms part of the region involved in specific protein/lipid interactions that favours homogeneous dystrophin distribution along the membrane39. Inspection of the 77 remaining 3′ss did not reveal any other NAGNAG tandem acceptor motifs in the DMD gene.
In conclusion, this study provides the first RNA-Seq-based reference of DMD splicing pattern in normal muscle. The strategy used allows the analysis of the whole 11.3 kb-cDNA sequence all at once that may prove useful for mutational analysis or monitoring the impact of splicing interventions on transcript structure in patients with Duchenne muscular dystrophy. Undesirable alternative splicing events may impact the outcome of exon skipping or trans-splicing approaches. Unlike other isoforms26,27,40, expression of the DMD gene is tightly regulated in skeletal muscle to preserve the production of a full-length transcript and a 427 kDa dystrophin protein, even if some protein domains are considered to be functionally dispensable41. The DMD gene represents a paradigm for extreme splicing conditions that illustrates perfectly how accurate the process of splice site selection shoud be to include the 79 exons into the mature transcript concomitantly with the repression of near perfect pseudoexons or cryptic 3′ss activation. A large piece of work remains to be done to elucidate the splicing regulation in this huge gene, which will contribute to reveal new insights into basic splicing mechanisms.
Methods
Muscle samples
Two striated muscle tissue samples (n°20316 and n°27563) from healthy male individuals (aged 29 and 19, respectively) were obtained from the Myobank-AFM (Paris, France). Two additional control skeletal muscle total RNA samples from 20 year-old males were purchased from Clontech (ref #636534, lots #1404229A and #1406360A).
RNA extraction and cDNA synthesis
Total RNA was extracted from biopsies using the SV total RNA extraction kit (Promega) and RNA integrity was checked using the Agilent RNA 6000 Nano kit with Agilent 2100 bioanalyzer (Agilent Technologies). 700 ng of total RNA were used to synthesize template cDNA by reverse transcription with the Superscript® II (Thermo Scientific) and oligodT primers for RNA-Seq experiments.
Long Range (LR)-PCR
One fifth of the RT product was used as template to amplify the full-length DMD cDNA as a single long fragment (11.3 kb) by using the GoTaq® Long PCR Master Mix (Promega) and primers located in exon 1 of the muscle isoform (Dp427m) (5′-CTTTCCCCCTACAGGACTCAG-3′) and in the 3′UTR (5′-CCAAATCATCTGCCATGTGG-3′). Cycle parameters were programmed as 94 °C for 2 min, followed by the first 10 cycles of 93 °C for 15 s, 58 °C for 30 s, and 68 °C for 11 min and a 20 s auto-extension of elongation time for cycles 11–35. After verification of amplicon size by agarose gel electrophoresis, LR-PCR reactions were purified using Qiaquick PCR purification kit (Qiagen) and quantified using the NanoDrop 2000 spectrophotometer (Thermo Scientific).
Construction of 454 libraries and sequencing
For each library, around 1 μg of purified PCR product were fragmented by nebulization (2.1 bars of nitrogen for 1 min) and subjected to library preparation with the Rapid Library Preparation Kit (Roche) according to manufacturer’s instructions. The different samples were labeled via ligation of Multiplex Identifiers (MID) oligonucleotide adaptors to allow multiplexing. Three MID-containing libraries were quantified using the Qubit® Fluorometer and pooled in equimolar amounts into a single sample prior to emulsion PCR amplification and sequencing in parallel on the Roche GS Junior 454 sequencer giving a mean total number of 69,060 reads (387 bp mean length) per library. The data obtained were then sorted according to their tag sequences.
Bioinformatics
Raw reads were edited and filtered prior to analysis. A dedicated analysis pipeline was developed using the Galaxy framework (http://galaxyproject.org)42. First, relevant adapter sequences were removed with Cutadapt (v.1.3, default parameters), and quality-based trimming at the 3′ ends of reads was performed using the Qtrim tool (v.1.1, parameters: mean quality = 25, window size = 20, minimum read length = 40 nt). Cleaned reads were then mapped to the Human X chromosome reference sequence (hg19, UCSC) with STAR (v. 2.3, annotations from UCSC, parameters: -sjdbOverhang 29 -outFilterMismatchNoverLmax 0.05 -outSJfilterReads Unique, outFilterMultimapNmax 1)20. A script developed in-house was developed to annotate identified splice junctions, which can be obtained upon request. The output data is processed to obtain DMD mRNA coverage, as well as a list of identified splice junctions and their counts. Only new junctions covered by a minimum of 5 reads in at least two out of the four biological replicates were considered for further analysis. The Percent-Spliced-In (PSI) was calculated for each splicing event using intron-centric metrics43, with the SJPIPE pipeline, from the ipsa package (parameters: margin = 0, deltaSS = 0, mincount = 0, https://github.com/pervouchine/ipsa). For the comparison of the DMD-targeted versus publicly available total mRNA sequencing data from the Illumina Human Body Map project 2.0 (skeletal muscle tissue, 2 × 50 bp, GEO sample GSM759515), raw reads were mapped to the Human X chromosome sequence with Bowtie2 (v2.default parameters)44 and exon junctions detection and count was performed by TopHat245 (v2.0.2, Gene Model Annotations option: chrX_GTF downloaded from UCSC). Two different splice site prediction algorithms were used for computational scoring of 5′ and 3′ splice sites: the Human Splicing Finder tool (http://www.umd.be/HSF3/), which uses the weight matrix model46 and the MaxEntScan (MES) (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html) based on the maximum entropy principle47.
Experimental validation of Alternative Splicing Events
Independent RT experiments were performed by using random primers for experimental validation. 1.5 μL of cDNA was used as template for PCR amplification in a 25 μL total volume with the Taq DNA Polymerase (New Englands Biolabs) and primers hybridizing to upstream and downstream exons (sequences available upon request). For conventional RT-PCR, the 30 cycles-amplified products were separated on 1.8% agarose gels and spliced products were quantified with the Quantity One (v.4.6.9) software (Bio-Rad). For semi-quantitative PCR, fluorescein-labeled forward primers were used and 26 cycles were done. Capillary electrophoresis analysis was performed using 1 μl of PCR product added to 18 μl of formamide and 0.5 μl of ROX 400 HD fluorescent size standards (Applied Biosystems). Amplified products were separated on an ABI 3130 XL DNA analyzer and the peaks areas were measured by the GeneMapper v4.0 software. Ratios of splicing isoforms were determined as the peak area of one specific isoform divided by the total peak areas for the two or three detected isoforms. Data represent the mean +/−SD of at least three independent assays.
Additional Information
How to cite this article: Bougé, A.-L. et al. Targeted RNA-Seq profiling of splicing pattern in the DMD gene: exons are mostly constitutively spliced in human skeletal muscle. Sci. Rep. 7, 39094; doi: 10.1038/srep39094 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
28 March 2017
A correction has been published and is appended to both the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
Pan, Q., Shai, O., Lee, L. J., Frey, B. J. & Blencowe, B. J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415 (2008).
Raj, B. & Blencowe, B. J. Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 87, 14–27 (2015).
Llorian, M. & Smith, C. W. J. Decoding muscle alternative splicing. Curr. Opin. Genet. Dev. 21, 380–7 (2011).
Castle, J. C. et al. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 40, 1416–1425 (2008).
Muntoni, F., Torelli, S. & Ferlini, A. Review Dystrophin and mutations: one gene. several proteins, multiple phenotypes. Lancet, 44, 731–740 (2003).
Tennyson, C. N., Shi, Q. & Worton, R. G. Stability of the human dystrophin transcript in muscle. Nucleic Acids Res. 24, 3059–3064 (1996).
Surono, A., Takeshima, Y., Wibawa, T., Pramono, Z. A. & Matsuo, M. Six novel transcripts that remove a huge intron ranging from 250 to 800 kb are produced by alternative splicing of the 5′ region of the dystrophin gene in human skeletal muscle. Biochem. Biophys. Res. Commun. 239, 895–899 (1997).
Sironi, M. et al. The dystrophin gene is alternatively spliced throughout its coding sequence. FEBS Lett. 517, 163–166 (2002).
Feener, C. A., Koenig, M. & Kunkel, M. Alternative splicing of human dystrophin mRNA generates isoforms at the carboxy terminus. Nature, 338, 509–511 (1989).
Zhang, Z. et al. Identification of seven novel cryptic exons embedded in the dystrophin gene and characterization of 14 cryptic dystrophin exons. J. Hum. Genet. 52, 607–17 (2007).
Guiraud, S., Chen, H., Burns, D. T. & Davies, K. E. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Exp. Physiol. 100, 1458–1467 (2015).
Miro, J. et al. FUBP1: a new protagonist in splicing regulation of the DMD gene. Nucleic Acids Res. 43, 2378–2389 (2015).
Disset, A. et al. An exon skipping-associated nonsense mutation in the dystrophin gene uncovers a complex interplay between multiple antagonistic splicing elements. Hum. Mol. Genet. 15, 999–1013 (2006).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Anders, S., Reyes, A. & Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017 (2012).
Halvardson, J., Zaghlool, A. & Feuk, L. Exome RNA sequencing reveals rare and novel alternative transcripts. Nucleic Acids Res. 41, 1–10 (2013).
Hooper, J. E. A survey of software for genome-wide discovery of differential splicing in RNA-Seq data. Hum. Genomics 8, 3 (2014).
Florea, L., Song, L. & Salzberg, S. L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research, 2, 188 (2013).
Nagalakshmi, U., Waern, K. & Snyder, M. RNA-Seq: A Method for Comprehensive Transcriptome Analysis. Curr. Protoc. Mol. Biol. 4, 1–13 (2010).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21 (2013).
Roberts, R. G., Bentley, D. R. & Bobrow, M. Infidelity in the structure of ectopic transcripts: a novel exon in lymphocyte dystrophin transcripts. Hum. Mutat. 2, 293–299 (1993).
Flanigan, K. M. et al. Nonsense Mutation-Associated Becker Muscular Dystrophy: interplay between exon definition and splicing regulatory elements within the DMD Gene. Hum Mutat 32, 299–308 (2011).
Tress, M. L. et al. The implications of alternative splicing in the ENCODE protein complement. Proc Natl Acad Sci USA 104, 5495–5500 (2007).
Thanh, L. T., Nguyen, T. M., Helliwell, T. R. & Morris, G. E. Characterization of revertant muscle fibers in Duchenne muscular dystrophy, using exon-specific monoclonal antibodies against dystrophin. Am J Hum Genet. 56, 725–731 (1995).
Lu, Q. L., Morris, G. E., Wilton, S. D., Ly, T., Artem’yeva, O. V., Strong, P. & Partridge T. A. Massive idiosyncratic exon skipping corrects the nonsense mutation in dystrophic mouse muscle and produces functional revertant fibers by clonal expansion. J Cell Biol. 148, 985–996 (2000).
Bies, R. D. et al. Human and murine dystrophin mRNA transcripts are differentially expressed during skeletal muscle, heart, and brain development. Nucleic Acids Res. 20, 1725–1731 (1992).
Austin, R. C., Morris, G. E., Howard, P. L., Klamut. H. J. & Ray, P. N. Expression and synthesis of alternatively spliced variants of Dp71 in adult human brain. Neuromuscul Disord. 10, 187–193 (2000).
Ellis, J. D. et al. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol Cell. 46, 884–892 (2012).
Le Rumeur, E., Winder, S. & Hubert, J. F. Dystrophin: More than just the sum of its parts. Biochim Biophys Acta. 1804, 1713–1722 (2010).
Lederfein, D. et al. A 71-kilodalton protein is a major product of the Duchenne muscular dystrophy gene in brain and other nonmuscle tissues. Proc Natl Acad Sci USA 89, 5346–5350 (1992).
Rau, F. et al. Abnormal splicing switch of DMD’s penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat Commun. 6, 7205 (2015).
Nishida, A., Minegishi, M., Takeuchi, A., Awano, H., Niba, E. T. & Matsuo, M. Neuronal SH-SY5Y cells use the C-dystrophin promoter coupled with exon 78 skipping and display multiple patterns of alternative splicing including two intronic insertion events. Hum Genet. 134, 993–1001 (2015).
Irimia, M. et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159, 1511–1523 (2014).
Li, Y., Sanchez-Pulido, L., Haerty, W. & Ponting, C. P. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 25, 1–13 (2015).
Gazzoli, I. et al. Non-sequential and multi-step splicing of the dystrophin transcript. RNA Biol. 13, 290–305 (2016).
Koren, E., Lev-Maor, G. & Ast, G. The Emergence of Alternative 3′ and 5′ Splice Site Exons from Constitutive Exons. PLoS Comput Biol. 3, e95 (2007).
Hiller, M. & Platzer, M. Widespread and subtle: alternative splicing at short-distance tandem sites. Trends Gene. 24, 246–255 (2008).
Yan, X., Sablok, G., Feng, G., Ma, J., Zhao, H. & Sun, X. nagnag: Identification and quantification of NAGNAG alternative splicing using RNA-Seq data. FEBS Lett. 589, 1766–1770 (2015).
Ameziane-Le Hir, S. et al. Cholesterol favors the anchorage of human dystrophin repeats 16 to 21 in membrane at physiological surface pressure. Biochim Biophys Acta 1838, 1266–1273 (2014).
Lidov, H. G. & Kunkel, L. M. Dp140: alternatively spliced isoforms in brain and kidney. Genomics 45, 132–139 (1997).
Mirabella, M., Galluzzi, G., Manfredi, G., Bertini, E., Ricci, E., De Leo, R., Tonali, P. & Servidei, S. Giant dystrophin deletion associated with congenital cataract and mild muscular dystrophy. Neurology 51, 592–595 (1998).
Blankenberg, D. et al. Galaxy, a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. 18, 1199–1216 (2010).
Pervouchine, D. D., Knowles, D. G. & Guigó, R. Intron-centric estimation of alternative splicing from RNA-seq data. Bioinformatics 29, 273–274 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Desmet, F. O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67 (2009).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394 (2004).
Acknowledgements
This work was supported by the Association Française contre les Myopathies (AFM-Téléthon) (grant number 18413 to STG). ALB received a post-doctoral fellowship from University of Montpellier (UM), France. We would like to thank the AFM tissue bank for research (MYOBANK-AFM) for generously providing normal human muscle biopsies.
Author information
Authors and Affiliations
Contributions
A.L.B. contributed to the design of the study, carried out experiments, analyzed and interpreted data, performed bioinformatics analysis, and helped draft the manuscript. E.M. contributed to the design of the study, carried out experiments, and analyzed data. E.B. implemented the bioinformatics analytical pipeline. J.M. participated in experiments and data analysis, J.V. and M.T. participated in the design of the study, M.K. participated in the interpretation of the data. E.M., E.B., J.M., J.V., M.T. and M.K. critically revised the manuscript. M.C. and S.T.G. conceived the study and participated in its design and revision of the manuscript. S.T.G. further coordinated and supervised the project, analyzed the data, and drafted the manuscript. All authors read and approved the final document.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bougé, AL., Murauer, E., Beyne, E. et al. Targeted RNA-Seq profiling of splicing pattern in the DMD gene: exons are mostly constitutively spliced in human skeletal muscle. Sci Rep 7, 39094 (2017). https://doi.org/10.1038/srep39094
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39094
This article is cited by
-
The exon junction complex is required for DMD gene splicing fidelity and myogenic differentiation
Cellular and Molecular Life Sciences (2024)
-
Nucleotides in both donor and acceptor splice sites are responsible for choice in NAGNAG tandem splice sites
Cellular and Molecular Life Sciences (2021)
-
The complexity of titin splicing pattern in human adult skeletal muscles
Skeletal Muscle (2018)
-
Analysis of extracellular mRNA in human urine reveals splice variant biomarkers of muscular dystrophies
Nature Communications (2018)
-
DMD transcripts in CRL-2061 rhabdomyosarcoma cells show high levels of intron retention by intron-specific PCR amplification
Cancer Cell International (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
