
Abstract
Heterozygous germline mutations in the human mismatch repair (MMR) genes MLH1, PMS2, MSH2 and MSH6 predispose to the hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Biallelic mutations in these genes have been reported for a limited number of cases resulting in hematological malignancies, brain tumors and gastrointestinal tumors early in childhood. These tumor phenotypes are frequently associated with café-au-lait spots (CALS), one of the clinical hallmarks of neurofibromatosis type 1 (NF1). We report the first case of compound heterozygosity for two MSH6 mutations resulting in a nonconservative amino-acid change of a conserved residue and in a premature stop codon in a patient who developed rectal and endometrial cancer at ages 19 and 24 years, respectively, and presented few CALS in a single body segment. Immunohistochemistry and Western blotting revealed only residual expression of the MSH6 protein in the normal cells. The disease history resembles the HNPCC phenotype rather than a phenotype associated with biallelic MMR gene mutations. Therefore, we assume that one or both mutations abolish protein function only partially, further supported by the parents, which are both carriers of one of the mutations each, and not affected by the disease at ages 57 and 58 years. Our data suggest considering biallelic mutations in MMR genes for patients who develop HNPCC-associated tumors at an unusually young age of onset, even without hematological or brain malignancies.
Similar content being viewed by others

Introduction
Heterozygous constitutional mutations of the mismatch repair (MMR) genes MSH2, MSH6, MLH1 and PMS2 predispose to hereditary nonpolyposis colorectal cancer (HNPCC, MIM 114500), an autosomal dominant cancer-susceptibility syndrome with high penetrance.1, 2 Mutation carriers are at high risk to develop colorectal (CRC) and endometrial cancer and are at increased risk to develop cancer of small bowel, stomach, ovary, ureter and hepatobiliary tract.3 A hallmark of these malignancies is contraction/expansion of simple sequence motifs termed microsatellite instability (MSI).4 Mutations in MSH6 have been shown to account for 10–15% of all MMR gene mutations identified in families suspected of HNPCC.5, 6 When compared to MSH2 and MLH1, tumors occurring in MSH6 mutation carriers (MIM no. 600678; GenBank accession: AH005068) are characterized by a lower frequency of instability at dinucleotide repeat markers.6 Clinically, these patients have a 10 years later age of tumor onset and a lower incidence of colorectal cancer, along with an increased incidence of endometrial cancer and other malignancies.5, 6, 7, 8, 9
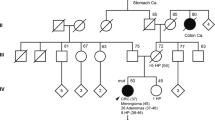
Biallelic MMR gene mutations have been reported for one or more patients of 14 families (2 × MSH2, 6 × MLH1, 4 × PMS2, 2 × MSH6).10, 11, 12 A total of 11 homozygous mutations were present resulting from proven or suspected consanguineous mating in most of the families. Compound heterozygous mutations have been shown once for MSH2 and twice for PMS2. In contrast to heterozygous MMR gene mutations, biallelic constitutional inactivation has been reported to result in the development of brain and colon tumors early in life, which is reminiscent of Turcot syndrome.13 Analogous to mouse models deficient in MMR, patients are at high risk to develop lymphomas, leukemia and gastrointestinal tumors early in life. In addition, they showed features of neurofibromatosis type 1 (NF1) such as café-au-lait spots (CALS, most patients), axillary freckling, Lisch nodules and neurofibromas (some patients), and tibial pseudarthrosis (one patient). Most patients with biallelic mutations in MLH1 and MSH2 died from hematological malignancies or brain tumors during the first decade of life.14, 15, 16, 17, 18 An exception constitutes the homozygous Arg687Trp mutation of MLH1, which was associated with colon and duodenal cancer at ages 9 and 11 years, respectively.19 In contrast, patients with biallelic mutations of PMS2 and MSH6 developed HNPCC-associated tumors (colon, rectum, duodenum, ovary) in addition to hematological malignancies and brain tumors, and tumor onset occurred primarily during the second and early third decade of life.10, 13, 20, 21, 22 This is the first report of a patient compound heterozygous for two MSH6 mutations who lacks hematological malignancies and brain tumors.
Materials and methods
Patient data and material
The female patient HD3 had a rectal cancer diagnosed at age 19 years and endometrial cancer diagnosed at age 24 years, and is currently disease free at age 29 years after undergoing appropriate surgical therapy. She was recruited through the Bethesda guidelines (B2: individuals with two HNPCC-related cancers and B4: individuals with colorectal or endometrial cancer diagnosed at age <45 years)23 and was subsequently offered HNPCC diagnostics. The only recorded malignancy among her relatives was a rectal carcinoma affecting the paternal grand mother at age 73 years. Written informed consent was obtained from all family members investigated. The study was approved by the local ethics committee.
T cells were isolated from peripheral blood of the patient and her parents and activated using T-cell expander (Dynal, Oslo, Norway). Genomic DNA from peripheral blood and from T cells of the patient and her parents and from paraffin-embedded tumor tissue of the rectal carcinoma of the patient was extracted applying the QIAamp® blood and tissue kit (QIAGEN, Hilden, Germany). Messenger RNA was isolated from peripheral blood and from T cells of the patient and her parents applying the Quick-Prep Micro mRNA Purification Kit and reverse transcribed using the First-Strand cDNA Synthesis Kit (both Amersham Biosciences, Freiburg, Germany) according to manufacturer's protocol.
Immunohistochemistry
Immunohistochemical staining was performed for MLH1, PMS2, MSH2 and MSH6 on 5-μm-thick, formalin-fixed, paraffin-embedded sections from both tumors of the patient as described previously.24 Staining in the absence of the primary antibody served as negative controls. The normal staining pattern for all four antibodies was nuclear. The pattern of staining was examined by subjective assessment of two independent examiners (JP and DEA).
Western blotting
Cells used for Western blotting were grown in DMEM/F12 medium supplemented with 10% fetal calf serum (PAA, Cölbe, Germany). As controls, the colorectal carcinoma cell lines HCT116 (MLH1-deficient) and DLD-1 (MSH6-deficient) as well as the human B-cell precursor leukemia cell line NALM6 (MSH2-deficient) (all DSMZ, Braunschweig, Germany) and two B-cell lines from different individuals were used. Cell lysates were prepared under denaturing conditions, proteins were separated on denaturing 4–16% precast gradient SDS-polyacrylamide gels (Invitrogen, Karlsruhe, Germany) and blotted onto nitrocellulose membranes. After blocking in 5% skimmed milk in TBS/Tween for 1 h at room temperature, membranes were probed with anti-MLH1 (Clone G168–15), anti-MSH2 (Clone 27), or anti-MSH6 (Clone 44; all antibodies were from BD Biosciences, Heidelberg, Germany). As a protein loading control, blots were also probed with anti-actin at 1:5000 (Oncogene, Schwalbach, Germany). As secondary antibody, we used horseradish peroxidase-conjugated anti-mouse Ig (1:1000, BD). All primary and secondary antibodies were diluted in blocking solution (5% skimmed milk powder, TBS/Tween) and were allowed to incubate for 1 h at room temperature. The immune complexes were detected using the ECL-Kit (Amersham-Pharmacia, Freiburg, Germany).
Molecular analyses
Analysis for microsatellite instability (MSI) in tumor tissue was performed applying six mono- and nine polynucleotide repeat markers by multiplex amplification in five panels as described previously.24 The markers include the National Cancer Institute/International Collaborative Group on HNPCC (NCI/ICG-HNPCC) reference marker panel for the evaluation of MSI in colorectal cancer (BAT25, BAT26, D2S123, D5S346, D17S250).25
For mutation analysis, the entire coding sequence and flanking intronic regions of MSH6 and MSH2 were amplified and sequenced from genomic DNA, as described previously.26 To examine mutations at the transcript level, RT-PCR analysis was performed using a sense primer located in exon 4 (GTE4S8: CGGAGGGATGTATCATTGAAG) and an antisense primer located in the 3′ end of the gene (GTE10cA1: CCACCTTTGTCAGAAGTCAA).
Allele assignment of the two mutations was performed by amplification of an approximately 3.6 kb fragment containing both mutation sites (sense primer GTE5S located in intron 4: TATTTTGATGGGGGAGATCG and antisense primer GTE8del2A located in intron 9: ATAGTGCATCATCCCTTCCC) applying the Expand Long Template PCR System (Roche Diagnostics, Mannheim, Germany) from genomic DNA and subsequent cloning into the plasmid vector pCR®2.1–TOPO® using the TOPO TA Cloning® Kit (Invitrogen, Karlsruhe, Germany). After transfection of competent bacteria cells (DH5α) and overnight cultivation, 20 single clones were grown, plasmid DNA was isolated and subsequently inserts were sequenced from both ends.
Results
The female patient was recruited as part of our routine screening for potential HNPCC families/patients through the Bethesda guidelines. Microsatellite analysis revealed instability at 4/6 mono- and 2/8 polynucleotide markers for the rectal tumor. Initial results on immunohistochemistry of the rectal tumor were: no staining for MSH6, weak staining for MSH2 and normal staining for MLH1. In addition, tumor-infiltrating lymphocytes did not stain for MSH6, which prompted us to conclude that staining of MSH6 was not meaningful in this tumor. Sequence analysis of MSH2 revealed no mutation, whereas a C-to-T transition at nucleotide 3226 of the MSH6 gene was identified.6 The mutation results in the substitution of cysteine for arginine (amino acid 1076, exon 5), leading to a nonconservative change from a basic to a polar, uncharged residue. Arg1076 is conserved for the interdomain interaction among a wide variety of mutS homologs ranging from human to bacteria.27 Therefore, this mutation may have a role in carcinogenesis.
To further clarify the expression pattern of MSH6 in normal cells, immunohistochemical analysis was repeated with normal colonic mucosa and with additional specimens of the rectal carcinoma, revealing lack of MSH6 expression in tumor cells, in normal crypt cells and in tumor-infiltrating lymphocytes (Figure 1). Absence of MMR protein expression in normal and tumor cells occurs occasionally, likely due to insufficient conservation of the paraffin-embedded tumor specimen and is mirrored by failure of staining of other proteins. The absence of MSH6 staining in tumor and normal cells, accompanied by normal staining of these proteins in other specimens in the same immunohistochemical series, indicate that the patient is completely or almost completely deficient for MSH6. This result was confirmed by Western blotting where T-cell extracts showed no signal for MSH6 using conventional protocols and a residual expression with increased protein loading and extended exposure time (Figure 2). We cannot exclude some dominant negative effect of the missense mutation on protein expression as cells of the mother seem to have a weaker expression of MSH6 than those of the father. Alternatively, it may simply represent different levels of activation and/or proliferation of the peripheral blood cells. Moreover, the mutation in exon 5 remains heterozygous in microdissected tumor DNA of two different specimens of the rectal tumor, basically excluding a second, somatic mutation in the form of allelic loss.

Expression of mismatch repair proteins, analyzed by immunostaining. (a–c) Tumor sections of patient HD3, harboring biallelic MSH6 germline mutations, immunostained with MSH6-, MSH2- and MLH1 antibodies, respectively. There is no MSH6 staining in tumor and infiltrating normal cells, whereas MSH2 is expressed weakly and MLH1 normally. (d and e) A different tumor specimen stained for MSH6 and MSH2 in the same series of immunohistochemical analyses serves as a positive control. (f–h) Normal colonic mucosa of the patient also lacks MSH6 expression, whereas MSH2 and MLH1 are expressed in normal cells. Original magnification: × 200.

Expression of mismatch repair proteins, analyzed by immunoblotting. (a) Western blotting revealed loss of MSH6 expression in normal T cells of patient HD3. Normal T cells of the parents express MSH6, albeit at a reduced level. The colorectal carcinoma cell lines HCT116 (MLH1-deficient) and DLD-1 (MSH6-deficient) as well as the human B-cell precursor leukemia cell line NALM6 (MSH2-deficient) and B-cell lines from two other individuals were used as controls. (b) To be able to finally decide whether there is a strong reduction or complete loss of MSH6 expression in normal cells of the patient, we repeated the Western blot analysis aiming at maximum sensitivity. Double amounts of protein (approximately 20 μg) were loaded for the patient and her parents, the incubation time for MSH6 antibody was extended from 1 h to overnight and the time of exposure was prolonged from 3 to 30 min.
These results prompted us to evaluate the complete MSH6 sequence. Sequencing of the coding region of MSH6 including the flanking intronic regions and the promoter revealed a second C-to-T transition affecting nucleotide 3991, which changes codon 1331 (exon 9) from arginine to stop codon and is predicted to remove the last 30 residues from the protein. Cloning and sequencing of a PCR-amplified fragment containing both mutation sites assigned the mutations to separate alleles. Compound heterozygosity was verified upon availability of samples from the parents, whereby the nonsense mutation was inherited from the father and the missense mutation from the mother, both of which are healthy at ages 58 and 57 years, respectively.
RT-PCR from mRNA of the three family members and controls amplified a shortened transcript in addition to the full-length transcript, exclusively from the patient and her father, which both harbor the nonsense mutation in exon 9 (Figure 3). Sequence analysis of the cDNA amplicons revealed skipping of exon 9 giving rise to the shorter transcript. In addition, the patient's shorter transcript was wild type at the site of the missense mutation in exon 5, confirming that the skipping of exon 9 exclusively occurred in the allele harboring the nonsense mutation in exon 9. Both mutations were seen from the full-length transcript of the patient indicating that the nonsense mutation in exon 9 only partially results in skipping of exon 9, and that the allele harboring the missense mutation in exon 5 is transcribed into mRNA.

RT-PCR analysis from normal cells of the patient and her parents. RT-PCR analysis from T cells of the patient and her parents resulted in a shortened transcript from the patient and the father (both harboring the nonsense mutation in exon 9) in addition to the transcript of wild-type size. Sequencing of all fragments revealed skipping of exon 9 for the shortened transcripts, retention of heterozygosity at the exon 9 mutation site in the wild-type-sized transcripts from the patient and the father, and wild type for the exon 5 mutation site for the shortened transcript of the patient. The wild-type-sized transcripts from the patient and the mother were both heterozygous at the exon 5 mutation site. Therefore, skipping of exon 9 is a consequence of the nonsense mutation within this exon and both mutations do not or only partially affect stability of respective transcripts.
Discussion
Homozygosity for a protein-truncating mutation in MSH6 has been reported as associated with a strong phenotype in two affected siblings, one of whom was affected by lymphoma and CRC diagnosed at ages 5 and 8 years, respectively, and the other by glioblastoma multiforme diagnosed at age 8 years also showing CALS and axillary freckling.28 Similar to the patient described here, parents were not affected and there was only one distant relative with CRC. Menko et al10 reported a child with oligodendroglioma at age 10 years and rectosigmoid cancer at age 12 years along with multiple CALS. The patient was homozygous for a 3 bp deletion in MSH6 due to consanguineous mating. The mutation results in a two-by-one replacement of nonconserved amino acids and was not associated with loss of protein expression in normal cells. In contrast to both these and the majority of the other reported cases of biallelic MMR gene mutations, our patient had no hematological malignancy or brain tumor. Like the majority of reported cases, the patient had CALS, whereby the segmental pattern may be related to a diminished rather than a completely abolished MMR capacity (Figure 4). She had also suffered from Lupus erythematosus, although it is not known whether this disease condition is associated with MMR deficiency. Her disease history resembles a strong expression of the HNPCC phenotype but is milder than the phenotype described so far for biallelic MMR gene mutations. This may be explained by the potential hypomorphic nature (abolishing protein function only partailly) of one or both mutations. Whereas no detectable MSH6 expression was seen in the immunohistochemistry, Western blotting revealed a faint signal from normal cells of the patient. Therefore, we cannot exclude a residual activity of the MSH6 protein. The transcript with the missense mutation in exon 5 and the transcript without exon 9 result in proteins with a changed amino acid, conserved for interdomain interaction, and one which lacks amino acids conserved for heterodimerization, respectively.27 Both the lack of stable protein conformation and the lack of heterodimerization are likely to result in protein degradation. In contrast, the transcript of the paternal allele (nonsense mutation in exon 9) with retention of exon 9 is predicted to result in a protein lacking the last 30 amino acids, which are not conserved among MutS homologs. This transcript may be responsible for the residual expression of MSH6. Moreover, the two reported cases of biallelic MMR gene mutations nonassociated with hematological malignancies or brain tumors in early childhood constitute homozygous missense mutations (both affecting MLH1), whereby partially MMR function was observed for one of these mutations.11, 19

(a, b) Café-au-lait spots in the patient. Café-au-lait spots in patient HD3 are restricted to the area of the right lower costal arch.
MSH6 mutations are associated with an approximately 10 years later age of disease onset and a lower incidence of colorectal cancer compared to MLH1 or MSH2 mutations.5, 6, 7, 8, 9 Therefore, families with MSH6 mutations are less completely enrolled by the usually applied selection criteria for testing on MMR gene mutations like the Amsterdam criteria and the Bethesda guidelines. Assuming the participation of hypomorphic mutations resulting in a depleted rather than an abolished protein function, the phenotype associated with biallelic MMR gene mutations may resemble the spectrum of HNPCC-associated tumors at young age without hematological and brain malignancies. Indeed, we have identified in a different patient affected with colon cancer at age 31 years two MSH6 mutations (p.C765W and p.V878A), proven to be biallelic (data not shown). The patient's mother, carrier of p.C765W, was affected with endometrial cancer at age 60 years. Two different mutations of the corresponding conserved residue of MSH2 have been reported for HNPCC patients in the ICG-HNPCC mutation databank (http://www.insight-group.org). Amino acid change p.V878A does not affect a conserved residue, but has been identified in several patients suspected of HNPCC along with absence in normal control populations.5, 7
An unidentified second mutation in a family will leave family members tested negative for the identified first mutation with a deceptive certainty about their disease risk. Therefore, molecular diagnostics of MMR genes should comprise complete mutation analysis for patients with an unusually young age of disease onset according to the affected gene. In addition, aberrant protein expression in normal tissue and HNPCC-associated tumors in both parental branches may indicate a missed second mutation in HNPCC families. Moreover, as currently applied methods do not detect all possible mutations, relatives of such patients may not be excluded from the surveillance program if only one mutation was identified. More stringent screening protocols have to be considered in cases with biallelic mutations, while less strict protocols on surveillance may be considered for relatives upon evidence of lower penetrance for a certain mutation.
Accession codes
References
de la Chapelle A : Genetic predisposition to colorectal cancer. Nat Rev Cancer 2004; 4: 769–780.
Peltomaki P, Vasen HF : Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 1997; 113: 1146–1158.
Lynch HT, Smyrk TC, Watson P et al: Genetics, natural history, tumor spectrum, and pathology of hereditary nonpolyposis colorectal cancer: an updated review. Gastroenterology 1993; 104: 1149–1535.
Aaltonen LA, Peltomaki P, Leach FS et al: Clues to the pathogenesis of familial colorectal cancer. Science 1993; 260: 812–816.
Wijnen J, de Leeuw W, Vasen H et al: Familial endometrial cancer in female carriers of MSH6 germline mutations. Nat Genet 1999; 23: 142–144.
Plaschke J, Engel C, Kruger S et al: Lower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 or MSH2 mutations: the German Hereditary Nonpolyposis Colorectal Cancer Consortium. J Clin Oncol 2004; 22: 4486–4494.
Berends MJ, Wu Y, Sijmons RH et al: Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am J Hum Genet 2002; 70: 26–37.
Peterlongo P, Nafa K, Lerman GS et al: MSH6 germline mutations are rare in colorectal cancer families. Int J Cancer 2003; 107: 571–579.
Hendriks YM, Wagner A, Morreau H et al: Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology 2004; 127: 17–25.
Menko FH, Kaspers GL, Meijer GA, Claes K, Van Hagen JM, Gille JJ : A Homozygous MSH6 mutation in a child with cafe-au-lait spots, oligodendroglioma and rectal cancer. Fam Cancer 2004; 3: 123–127.
Raevaara TE, Gerdes AM, Lonnqvist KE et al: HNPCC mutation MLH1 P648S makes the functional protein unstable, and homozygosity predisposes to mild neurofibromatosis type 1. Genes Chrom Cancer 2004; 40: 261–265.
Rey JM, Noruzinia M, Brouillet JP, Sarda P, Maudelonde T, Pujol P : Six novel heterozygous MLH1, MSH2, and MSH6 and one homozygous MLH1 germline mutations in hereditary nonpolyposis colorectal cancer. Cancer Genet Cytogenet 2004; 155: 149–151.
Hamilton SR, Liu B, Parsons RE et al: The molecular basis of Turcot's syndrome. N Engl J Med 1995; 332: 839–847.
Wang Q, Lasset C, Desseigne F et al: Neurofibromatosis and early onset of cancers in hMLH1-deficient children. Cancer Res 1999; 59: 294–297.
Ricciardone MD, Ozcelik T, Cevher B et al: Human MLH1 deficiency predisposes to hematological malignancy and neurofibromatosis type 1. Cancer Res 1999; 59: 290–293.
Vilkki S, Tsao JL, Loukola A et al: Extensive somatic microsatellite mutations in normal human tissue. Cancer Res 2001; 61: 4541–4544.
Whiteside D, McLeod R, Graham G et al: A homozygous germ-line mutation in the human MSH2 gene predisposes to hematological malignancy and multiple cafe-au-lait spots. Cancer Res 2002; 62: 359–362.
Bougeard G, Charbonnier F, Moerman A et al: Early onset brain tumor and lymphoma in MSH2-deficient children. Am J Hum Genet 2003; 72: 213–216.
Gallinger S, Aronson M, Shayan K et al: Gastrointestinal cancers and neurofibromatosis type 1 features in children with a germline homozygous MLH1 mutation. Gastroenterology 2004; 126: 576–585.
De Rosa M, Fasano C, Panariello L et al: Evidence for a recessive inheritance of Turcot's syndrome caused by compound heterozygous mutations within the PMS2 gene. Oncogene 2000; 19: 1719–1723.
Trimbath JD, Petersen GM, Erdman SH, Ferre M, Luce MC, Giardiello FM : Cafe-au-lait spots and early onset colorectal neoplasia: a variant of HNPCC? Fam Cancer 2001; 1: 101–105.
De Vos M, Hayward BE, Picton S, Sheridan E, Bonthron DT : Novel PMS2 pseudogenes can conceal recessive mutations causing a distinctive childhood cancer syndrome. Am J Hum Genet 2004; 74: 954–964.
Rodriguez-Bigas MA, Boland CR, Hamilton SR et al: A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: Meeting highlights and Bethesda guidelines. J Natl Cancer Inst 1997; 89: 1758–1762.
Plaschke J, Krüger S, Pistorius S, Theissig F, Saeger HD, Schackert HK : Involvement of hMSH6 in the development of hereditary and sporadic colorectal cancer revealed by immunostaining is based on germline mutations, but rarely on somatic inactivation. Int J Cancer 2002; 97: 643–648.
Boland CR, Thibodeau SN, Hamilton SR et al: A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998; 58: 5248–5257.
Plaschke J, Kruppa C, Tischler R et al: Sequence analysis of the mismatch repair gene hMSH6 in the germline of patients with familial and sporadic colorectal cancer. Int J Cancer 2000; 85: 606–613.
Obmolova G, Ban C, Hsieh P, Yang W : Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 2000; 407: 703–710.
Hegde MR, Chong B, Blazo ME et al: A homozygous mutation in MSH6 causes Turcot syndrome. Clin Cancer Res 2005; 11: 4689–4693.
Acknowledgements
We thank Ms M Reichmann and Ms A Colditz for excellent technical assistance. This work was supported by the Deutsche Krebshilfe grant ‘Familiärer Darmkrebs’ (70-2367-Scha2).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Plaschke, J., Linnebacher, M., Kloor, M. et al. Compound heterozygosity for two MSH6 mutations in a patient with early onset of HNPCC-associated cancers, but without hematological malignancy and brain tumor. Eur J Hum Genet 14, 561–566 (2006). https://doi.org/10.1038/sj.ejhg.5201568
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201568
Keywords
This article is cited by
-
Heterogeneous constitutional mismatch repair deficiency with MSH6 missense mutation clinically benefits from pembrolizumab and regorafenib combination therapy: a case report and literature review
Hereditary Cancer in Clinical Practice (2021)
-
Report of the fifth meeting of the European Consortium 'Care for CMMRD' (C4CMMRD), Leiden, The Netherlands, July 6th 2019
Familial Cancer (2021)
-
Gastrointestinal Findings in the Largest Series of Patients With Hereditary Biallelic Mismatch Repair Deficiency Syndrome: Report from the International Consortium
American Journal of Gastroenterology (2016)
-
The Gastrointestinal Phenotype of Germline Biallelic Mismatch Repair Gene Mutations
American Journal of Gastroenterology (2010)
-
Functional characterization of rare missense mutations in MLH1 and MSH2 identified in Danish colorectal cancer patients
Familial Cancer (2009)
