
Abstract
Autosomal recessive ulcero-mutilating neuropathy with spastic paraplegia is a very rare disease since only few cases were described up to date. We report in this study a consanguineous Moroccan family with four affected males with this syndrome. The disease onset was in early infancy, with spastic paraplegia and sensory loss leading to mutilating acropathy. Electrophysiological studies revealed a severe axonal sensory neuropathy, magnetic resonance imaging ruled out compression of spinal cord and biological investigations showed decreased levels of Apo B, total cholesterol and triglycerides. A genomewide search was conducted in this family and linkage was found to chromosome 5p. Analysis of recombination events and LOD score calculation map the responsible gene in a 25 cM genetic interval between markers D5S2054 and D5S648. A maximum LOD score value of 3.92 was obtained for all markers located in this candidate interval. This study establishes the presence of a locus for autosomal recessive mutilating sensory neuropathy with spastic paraplegia on chromosome 5p15.31–14.1.
Similar content being viewed by others


Introduction
In the clinically and genetically heterogeneous group of the hereditary neuropathies, the peripheral nerve disorder can either be pure or associated with widespread neurological disorders.1 Syndrome combining mutilating hereditary sensory neuropathy and spastic paraplegia is a very rare disease which has been poorly documented in the literature. Indeed, only five families with autosomal dominant mode of inheritance,2, 3, 4 and four families in which the inheritance was likely to be autosomal recessive have been described.5, 6 Sporadic cases with similar features and normal parents have been also described in which the inheritance could be autosomal recessive or due to a de novo dominant mutation.5, 6, 7, 8 These so far described families have not led to the identification of the responsible genes neither for the dominant nor recessive forms.
In the present study, we report a consanguineous Moroccan family with four patients displaying an autosomal recessive mutilating hereditary sensory neuropathy with spastic paraplegia. A genomewide search was performed and evidence for linkage was found in chromosome 5p in a genetic interval of 25 cM. Biological investigation detected decreased levels of Apo B lipoprotein, cholesterol and triglycerides suggesting that the disease mechanism may be related to lipoprotein metabolism.
Patients and methods
Family
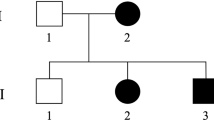
A consanguineous family of Moroccan origin (Figure 1) was examined in the Department of Neurology and Neurogenetics, Hôpital des Spécialités, Rabat, Morocco. Clinical and electrophysiological examinations were performed in 12 family members including four affected males, six at-risk relatives and the parents. All four affected individuals, with an age at exam of 34, 22, 19 and 7 years, have a similar clinical phenotype. The disease onset was in the early infancy and ranged from 1 to 5 years. Clinical examination revealed spasticity in walking with legs in form of X, increased tendon reflexes with ankle clonus, Babinsky sign, and discreet distal amyotrophy, whereas motor function was normal. Distal sensory loss for all modalities was observed in both upper and lower limbs particularly in foot. Deformities were present in hands and feet of two patients, and deep perforating ulcers were noted in fingers, toes and plantar depending on patient. The oldest patient IDR-004 presented scars of healed ulcers in hands and osteomyelitis of the feet leading to amputation of his two legs up to the thighs. During the disease course, the evolution of pyramidal symptoms was slow and of long duration, patients are still autonomous at age of exam; while the sensory neuropathy was very severe and developed rapidly with chronic mutilations and amputations. Cranial nerves, coordination, and cognitive functions were normal in all patients.

Haplotype reconstruction for chromosome 5p in pedigree IDR. Microsatellite markers are ordered according to the Marshfield genetic map from centromere (top) to telomere (bottom). The hatched area represents the common region of homozygosity for all affected individuals.
Motor conduction velocity of the median and the peroneal nerves was normal or slightly reduced, with ranges of 44–62.9 m/s (mean 51.22 m/s) for the median nerve and 37.2–44.4 m/s (mean 40.43 m/s) for the peroneal. Sensory action potentials (SNAPs) of the median nerve were not detectable in the oldest patient IDR-004; their amplitude value is reduced in patients IDR-008 and IDR-009 (7.5 and 3.4 mV, respectively) and normal in the youngest patients IDR-012 (24.6 mV), whereas NCVs values were 48, 45, and 46.1 m/s, respectively. SNAPs of the sural nerve were detectable only in the youngest patient IDR-012 with amplitude value of 8.4 mV and NCV value of 40 m/s. Motor and sensory recordings of peroneal and sural nerves were not applicable in patient IDR-004 since his two legs were amputated. These electrophysiological findings confirm a severe sensory axonal polyneuropathy predominantly in lower limbs. The exploration of the autonomic system in patient IDR-009 using ECG, deep breathing hand grip, valsalva and tilt tests revealed vagal hyperactivity with decreased α-sympathetic responses and increased β-sympathetic responses. Both parents and the other six siblings have normal clinical and neurophysiological investigations.
Magnetic resonance imaging exploration performed in patients IDR-008 and IDR-009 showed severe atrophy of the spinal cord. Laboratory investigations including routine hematology and biochemistry performed in patients IDR-008 and IDR-012 revealed decreased levels of Apo B lipoprotein (0.36 and 0.42 g/l, respectively; (0.70–1.30)), total cholesterol (1.09 and 1.34 g/l, respectively; (1.60–2)) and triglycerides (0.48 and 0.38 g/l, respectively; (0.60–1.60)), while Apo A lipoprotein levels were normal (1.12 and 1.37 g/l, respectively; (1.1–1.6)).
The detailed description of the disease phenotype will be reported elsewhere (Benormar et al, submitted).
Genetic linkage analysis
Blood samples from the 12 subjects were obtained after informed consent, and genomic DNA was extracted using standard procedures. A genomewide search was carried out using the ABI PRISM linkage mapping set version 2.5 from Applied BiosystemsR with a resolution of approximately 10 cM. Genotyping was performed with an automated capillary DNA sequencer ABI 310 and data were collected and analysed using the ABI GENESCAN (version 2.1) and GENOTYPER (version 2.0) software (Applied Biosystems, Foster City, CA, USA). Pairwise and multipoint LOD scores were calculated using the Allegro v1.2c2 software.
Results
We described a single family with four affected members and autosomal recessive pattern of inheritance. Since only males are affected, we first tested linkage with the 18 markers of the chromosome X panel. Pairwise lod scores were negative for all these markers (data not shown). After genotyping of 280 (73%) markers of the autosomal genome, linkage was established to chromosome 5p when positive lod scores were only obtained with markers D5S406, D5S630 and D5S416 (Table 1). Therefore, 13 additional markers spanning this region were tested in the pedigree and lod scores above threshold of 3 at θ=0.0 were obtained with several markers (Table 1). The most likely disease haplotype was constructed according to the Marshfield genetic map (Figure 1). Patient IDR-009 is homozygous for all the 16 markers tested, while a recombination event was observed in patient IDR-008 between D5S2054 and D5S1953 markers, placing the disease gene distal to D5S1953. The telomeric boundary of the genetic candidate interval is delimited by the recombination occurred in patient IDR-004 between D5S2074 and D5S648. The combined information given by these two recombinants place the disease gene in a 25 cM region between markers D5S2054 and D5S648. This result was confirmed by multipoint linkage analysis (Figure 2) which demonstrated that all markers in this candidate interval gave the same lod score value of Zmax=3.92.

Multipoint linkage analysis in family IDR. The genetic distances (in cM) are represented as on the Marshfield chromosome 5 map.
Discussion
We studied a Moroccan family with autosomal recessive mode of inheritance and four affected males displaying an ulcero-mutilating neuropathy with spastic paraplegia. Disease onset was in early infancy with spasticity in walking and a progressive severe sensory loss leading to chronic ulcerations in both upper and lower limbs. Electrophysiological studies revealed an axonal sensory neuropathy. As for the so far described families, the major clinical feature in our family was a progressive distal sensory neuropathy, whereas the spastic paraplegia was relatively mild.
Abnormal lipoprotein profile is not a common finding in autosomal recessive mutilating sensory neuropathy with spastic paraplegia, which suggests that the pathogenic mechanism in our family may be related to the lipoprotein metabolism. This unusual feature, together with atrophy of spinal cord and the disturbance of the peripheral autonomic system, differentiate our family from those of Thomas et al.6 The clinical picture in our family most likely corresponds to the ‘Cavanagh syndrome’.5
The genomewide search in this family resulted in the identification of the first locus for this rare and distinct clinical entity on the chromosome 5p15.31–14.1. Patients shared homozygosity for all markers between D5S2054 and D5S648, and multipoint LOD score analysis gave significant and positive values of 3.92 for all these markers, placing the disease gene in a 25 cM genetic interval between D5S2054 and D5S648 microsatellites. More than 90 genes and pseudogenes have been mapped (Human Genome Browser) in this candidate genetic interval. Possible candidate genes for this mutilating sensory neuropathy with spastic paraplegia syndrome, on the basis of their function, include the Semaphorin 5A gene (SEMA5A, OMIM 609297) which is a member of the semaphoring protein family involved in axonal guidance during neural development,9 and the brain-abundant signal protein membrane-attached 1 gene (BASP1, OMIM 605940), which is specifically expressed in nervous tissue. The most obvious functional candidate gene seems to be the Cct5 gene (ENSG00000150753) encoding the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (CCT complex) involved in folding actin and other cytosolic proteins.10 In deed, mutation in Sprague–Dawely rat Cct4 gene, which encode the delta subunit of the CCT complex, has been shown to be associated with mutilating sensory neuropathy;11 whereas mutation in the SPG13 gene encoding the mitochondrial chaperonin Hsp60 has been reported to cause hereditary spastic paraplegia.12 Screening of these three candidate genes (SEMA5, BASP1 and Cct5) and many others from the candidate interval is currently being performed.
In conclusion, we establish the presence of a locus for autosomal recessive mutilating sensory neuropathy with spastic paraplegia on chromosome 5p15.31–14.1 in a 25 cM genetic interval between markers D5S2054 and D5S648. Further refinements may reduce the number of positional candidate genes that need to be analyzed for the identification of mutations in order to identify the responsible gene.
Accession codes
References
Reilly MM, Hanna MG : Genetic neuromuscular disease. Neurol Pract 2002; 73: ii12–ii21.
Van Epps C, Kerr HD : Familial lumbosarcal syringomyelia. Radiology 1940; 35: 160–173.
Khalifeh RR, Zellweger H : Hereditary sensory neuropathy with spinal cord disease. Neurology 1963; 13: 405–411.
Koenig RH, Spiro AJ : Hereditary spastic paraplegia with sensory neuropathy. Dev Med Child Neurol 1970; 12: 576–581.
Cavanagh NP, Eames RA, Calvin RJ, Brett EM, Kelly RE : Hereditary sensory neuropathy with spastic paraplegia. Brain 1979; 102: 79–94.
Thomas PK, Misra VP, King HM et al: Autosomal recessive hereditary sensory neuropathy with spastic paraplegia. Brain 1994; 117: 651–659.
Tenembaum SN, Reisin RC, Taratuto AL, Fejerman N : Spastic paraplegia and sensory neuropathy. Muscle Nerve 1996; 19: 649–653.
Kherbaoui-Redouani L, Ploton D, Abely M et al: Hereditary sensory neuropathy with spastic paraplegia. Eur J Paediatr Neurol 2004; 8: 95–99.
Adams RH, Betz H, Puschel AW : A novel class of murine semaphorins with homology to thrombospondin is differentially expressed during early embryogenesis. Mech Dev 1996; 57: 33–45.
Yokota S, Yanagi H, Yura T, Kubota H : Cytosolic chaperonin-containing t-complex polypeptide 1 changes the content of a particular subunit species concomitant with substrate binding and folding activities during the cell cycle. Eur J Biochem 2001; 268: 4664–4673.
Lee MJ, Stephenson DA, Groves MJ et al: Hereditary sensory neuropathy is caused by a mutation in the delta subunit of the cytosolic chaperonin-contaning t-complex peptide-1 (Cct4) gene. Hum Mol Genet 2003; 12: 1917–1925.
Hansen JJ, Dürr A, Cournu I et al: Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the Mitochondrial chaperonin Hsp60. Am J Hum Genet 2002; 70: 1328–1332.
Acknowledgements
We thank all the family members for their cooperation throughout this study. This research was supported by the Presidency of the University Mohamed V – Souissi (Rabat, Morocco), and the Association Marocaine de Neurogénétique (Morocco). We are grateful for Genome Biotechnology SARL for their collaboration.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bouhouche, A., Benomar, A., Bouslam, N. et al. Autosomal recessive mutilating sensory neuropathy with spastic paraplegia maps to chromosome 5p15.31–14.1. Eur J Hum Genet 14, 249–252 (2006). https://doi.org/10.1038/sj.ejhg.5201537
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201537
Keywords
This article is cited by
-
A review of the genetic spectrum of hereditary spastic paraplegias, inherited neuropathies and spinal muscular atrophies in Africans
Orphanet Journal of Rare Diseases (2022)
-
Lipid chaperones and associated diseases: a group of chaperonopathies defining a new nosological entity with implications for medical research and practice
Cell Stress and Chaperones (2020)
-
Bridging human chaperonopathies and microbial chaperonins
Communications Biology (2019)
-
Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms
Acta Neuropathologica (2013)
-
Mechanisms of disease in hereditary sensory and autonomic neuropathies
Nature Reviews Neurology (2012)
