
Abstract
Four unrelated patients with glyceroluria ranging from 7 to 170 mmol/l were studied. The activity of glycerol kinase (GK) in cultured fibroblasts was determined with a specific enzyme assay and with two indirect methods, that is, incorporation into macromolecules of [14C] from [14C]glycerol and its oxidation to [14C]CO2. Exon amplification and RT-PCR were used to identify mutations. In patient 1, with low activity in all three assays, we identified a c.1194A>C (E398D) missense mutation. In patient 2 with a considerable activity of the GK enzyme (22% of reference), oxidation to [14C]CO2 (37%) and a high incorporation of [14C] into macromolecules (92%), we identified a c.182T>C (L61P) mutation that causes the enzyme to have a higher Km for glycerol (∼300 μ M) than normals (2–8 μ M). In patient 3, the GK activity estimated by the three different methods ranged from 16 to 22% of reference. Analysis of mRNA from the GK gene revealed three alternatively spliced transcripts. A mutation in intron 3 (g.16835G>A) resulted in an insertion of a cryptic exon between exon 2 or 3 and exon 4. Patient 4 with minor glyceroluria (7 mmol/l) and normal plasma glycerol concentration had normal activity with all three assay methods, thus excluding GK deficiency (GKD) as a cause of slight glyceroluria. To evaluate fully patients with glyceroluria, one needs to measure the GK activity and relate this and the clinical data to genetic findings. Residual enzyme activities in cultured fibroblasts can be found in GKD patients with severe clinical symptoms.
Similar content being viewed by others

Introduction
Glycerol kinase deficiency (GKD, MIM 307030) is an X-linked recessive enzyme defect characterized biochemically by hyperglycerolemia and glyceroluria. Isolated GKD can occur in patients with or without symptoms, mainly due to disturbed energy homeostasis associated with hyperketotic hypoglycemia.1 It has recently been shown that subjects with severe catabolic crises in childhood may become symptom free at adult age.2 Deletion of the GK locus together with other loci in the Xp21 region results in the enzyme deficiency in combination with adrenal hypoplasia congenita (AHC) and/or Duchenne muscular dystrophy (DMD).3 These patients may be mentally retarded and show dysmorphic features.4
In GKD patients, glycerol concentration in urine is usually >150 mmol/l (reference ⩽0.2 mmol/l) and in plasma it is ∼2–8 mmol/l, whereas normally it does not exceed 0.28 mmol/l.3 An elevated plasma glycerol concentration can erroneously result in an overestimation of plasma triglycerides known as pseudohypertriglyceridemia.5 The GK enzyme (EC 2.7.1.30; ATP:glycerol 3-phosphotransferase) activity can be assessed to confirm the diagnosis of GKD using either a specific enzyme assay,6 or indirect but sensitive assays that measure radio-labeled glycerol oxidation7 or incorporation of [14C]glycerol carbon in macromolecules of intact cultured fibroblasts.8 Until now, the activity of GK in GKD individuals has been <1–5% of control enzyme activity assayed by either assay in our laboratories,1,6 but higher levels of GK activity have been reported from other groups.9,10,11
We here describe four patients with glyceroluria between 7 and 178 mmol/l with a wide range of clinical symptoms and different levels of GK activity. We used mutation analysis to look for aberrations in the GK gene that could explain a significant residual GK activity. Kinetic analysis and predictions of the enzyme structure by molecular modeling were used to study the effects of the mutations.
Patients and methods
Clinical reports
Patient 1, a 4-year-old boy, born after 37 weeks of gestation with a birth weight of 2210 g. His mother had gestational diabetes. When the patient was 2 years old, he had periods of episodic vomiting, which led to the clinical and biochemical diagnosis of GKD. At 4.5 years, he showed mild asymmetric spasticity, hyperactivity, mild mental retardation, convergent strabismus, myopia and mild facial dysmorphism. Analysis of plasma triglyceride showed pseudohypertriglyceridemia 4 mmol/l (reference 0.4–1.2 mmol/l), and analysis of urine organic acids showed massive glyceroluria (170 mmol/l).
Patient 2, a 4-year-old boy, had two episodes of somnolence after vomiting necessitating hospital admission at the age of 2 years. Ketoacidosis was a prominent finding and selective screening for inborn errors of metabolism revealed glyceroluria (58 mmol/l) and glycerolemia (4 mmol/l). Plasma cortisol concentration and serum creatine kinase activity were normal. A presumptive diagnosis of isolated GKD was carried out. In an effort to estimate the duration of a safe fasting period, a diagnostic fasting test was performed at the age of 4 years.12 After 24 h of fasting, blood glucose was 3.6 mmol/l (controls 2.8–3.8 mmol/l),13 and there was a moderate metabolic acidosis (pH 7.31; base excess −8.9 mEq/l) with an impressive ketonemia and ketonuria. Plasma 3-hydroxybutyrate concentration at the end of the fasting test was 5.8 mmol/l (controls 1.7–3.2 mmol/l).13 The psychomotor development and growth were considered to be normal.
Patient 3, a male newborn, was born at 34 weeks of gestation with a birth weight of 1950 g and severe asphyxia. At 1 h after birth, a blood gas analysis showed respiratory acidosis. Electrolyte examination showed a normal blood sodium (133–139 mmol/l) and an elevated blood potassium level (9 mmol/l) suggestive of adrenal gland imbalance. Chest radiography suggested a respiratory distress syndrome complicated by a pneumothorax. Adrenal hypoplasia was suspected because of a deficiency of the fetal adrenal zone steroid 16-OH-pregnenolone (127.5 μg/24 h, reference: 194–1293 μg/24 h) as well as the absence of the cortisol metabolite tetrahydrocortisone in the urine.14 The activity of creatine kinase in serum was normal. Analysis of urine and plasma organic acids showed hyperglycerolemia (12 mmol/l) and glyceroluria (31 mmol/l). These findings suggested the diagnosis of complex GKD (AHC-GKD). The patient died on the fifth day of life because of circulatory collapse, before diagnostic testing for AHC could be performed.
Patient 4, a 9½year-old boy was born at a gestational age of 37 weeks with a birth weight of 2760 g. The pregnancy was complicated by premature rupture of the membrane. He was hospitalized for neonatal meningitis. By the age of 3 years, he was referred again because of a delayed development and hypotonia with a positive Gower's sign, and persistent unexplained microcytic anemia. The serum creatine kinase level was normal, and metabolic investigations revealed glyceroluria (7 mmol/l), whereas the glycerol levels in plasma were persistently normal.
Glycerol measurements
Glyceroluria was detected by selective screening of urinary organic acids by gas chromatography-mass spectrometry (GC-MS) of the trimethylsilyl derivatives. Glycerol was either extracted with ethyl acetate or by normal phase extraction.15 The quantitative determination of glycerol in urine was carried out by a triglyceride test kit, which measures glycerol after hydrolysis. Plasma glycerol was measured with an enzymatic method omitting triglyceride hydrolysis.
[14C]glycerol carbon incorporation into macromolecules
GK activity was indirectly assayed in intact fibroblasts by measurement of the incorporation of [14C]glycerol carbon into trichloroacetic acid (TCA)-precipitable material essentially as described by McCabe et al8 Fibroblasts were cultured in 35 mm dishes in Ham's F10 medium with 15% fetal calf serum (FCS) and antibiotics. Confluent cultures were incubated for 24 h, at 37°C in 1.5 ml F10 medium without FCS, supplemented with 1 μCi/ml of [U-14C]glycerol (171 mCi/mmol, Amersham). After harvesting with trypsin, cells were disrupted in 0.5 ml ice-cold 5% TCA and keeping the tubes on ice for 10 min and subsequent centrifugation precipitated macromolecular material. Following further treatment with cold 5% TCA, the final precipitates were dissolved in 100 μl of 1 mol/l NaOH. Radioactivity in each sample was measured by scintillation counting, while protein was quantitated using the bicinchoninic acid method. The incorporation of radioactivity from [U-14C]glycerol into macromolecular material was expressed as dpm [14C]/μg protein.
Glycerol oxidation to CO2
Confluent layers of fibroblasts in cell-culture flasks were trypsinized and the cells counted. Equal numbers of cells (300 000) were seeded into 25 cm2 cell-culture flasks with minimum essential medium Eagle with Earle's salts. When the cell monolayers were confluent, the flasks were washed twice with phosphate-buffered saline and 1 μCi of [U14C]glycerol (specific activity 143 mCi/mmol) in 1.2 ml Krebs Ringer phosphate buffer pH 7.4 was added to one 25 cm2 cell-culture flask for each incubation time used. The flasks were incubated at 37°C for 2–6 h. The reaction was stopped by adding 1.2 ml 10% TCA and CO2 was collected on a filter paper with 50 μl 1 mol/l hydroxide of Hyamine® 10-X in methanol, for an additional hour. The formed amount of [14C]CO2 was counted in a Beckman LS5000 in 2.5 ml methanol and 7.5 ml Ready Safe™ scintillation cocktail.7
GK activity measurements
GK activity was measured in fibroblasts after permeabilization by digitonin. The assay is based on the GK catalyzed conversion of [14C]glycerol to [14C]glycerol-3-phosphate, and the formed amount of [14C]glycerol-3-phosphate was determined by scintillation counting.6,7 The measurements were carried out in duplicate at two different protein concentrations between 0.5 and 3.3 g/l at a [14C]glycerol concentration of 45 μmol/l. The protein determination was performed on a cell suspension dissolved in 0.5 mol/l NaOH according to the method of Lowry et al16 Substrate saturation assay was performed as described for the enzymatic GK assay with 10 different glycerol concentrations from 1 to 110 μmol/l. Michaelis–Menten constants, Km and Vmax, were derived by Hanes–Woolf analysis.
Mutation analysis
Genomic – PCR
DNA was isolated from peripheral blood leukocytes and cultured fibroblasts according to established procedures. Genomic DNA of the index cases and one unrelated normal individual were used for mutation analysis. The entire protein-coding region of the GK gene was analyzed for mutations by sequencing the PCR-amplified exons. Of the 21 exons of the GK gene (Accession No. AJ252550-AJ252560, AJ252563-AJ252570), 19 were amplified using genomic primers as described previously.1,10,17 To analyze exons 10 and 11, the Expand™ High Fidelity PCR system (Boehringer Mannheim, Germany) was used to amplify the long DNA fragments comprising exons 8–11 according to the manufacturer's instruction as described previously.1 Sequencing was performed using the dRhodamine dye terminator cycle sequencing kit (Perkin-Elmer) and the products were analyzed on an ABI 377 sequencer (Applied Biosystems™, Foster City, CA, USA).
DNA sequence changes were confirmed by allele-specific oligonucleotide hybridization (ASOH). To hybridize duplicate dot blots of the appropriate genomic fragments, we used T4 polynucleotide kinase radio-labeled sequences;18 ASO-Normal (TGC ATT AGA AGC TGT TTG T, exon 14) and (AGG AAA TTC TAC ATT CTG T, exon 3) and ASO-Mutation (TGC ATT AGA CGC TGT TTG T, patient 1) and (AGG AAA TTC CAC ATT CTG T, patient 2).
To establish the AHC involvement, the DAX1 gene (Dosage-sensitive sex reversal – AHC critical region on the X-chromosome, gene 1) mutation analysis was performed in patient 3 essentially as described previously.19
Alterations found in the DNA sequence are compared to GenBank Sequence Accession No. L13943 (The Human Gene Mutation Database, HGMD®) and numbered according to published recommendations.20
RT-PCR
Total RNA was purified from fibroblasts with the QIAamp® RNA Blood kit (QIAGEN GmbH, Hilden, Germany) according to their protocol. The QIAGEN® One-Step RT-PCR kit was used for the amplification of the GK Xp21-specific mRNA transcript (EMBL X78211) with primers designed to amplify this active gene copy exclusively.21 The Xp21 GK gene was amplified as two overlapping fragments, a 667 bp fragment covering exons 1–8 and a 1190 bp fragment covering exons 8–19. Amplified fragments were separated on a 1.5% agarose gel, excised, purified and sequenced on an ABI 377 sequencer using their BigDye™ Terminator kit for the cycle-sequencing reaction. The g.16835G>A mutation (reference sequence NT_011757) found in DNA from patient 3 was confirmed by allele-specific PCR. Two PCR reactions were performed simultaneously with a sense primer detecting the normal allele (TCC TTT CTC CCT TGT TTT TCC G) or the mutated allele (TCC TTT CTC CCT TGT TTT TCC A) with an antisense primer (AGT TGT ACA GAT GGA AGT G). The sense primers were designed with an additional mismatch (marked in bold). PCR conditions were as described by the enzyme manufacturer with 200 ng DNA, 2.5 mmol/l MgCl2, 100 pmol primer and an annealing temperature of 57°C.
Results
Routine analysis of urinary organic acids as their trimethylsilyl derivates by GC-MS will not pick up the normal urine glycerol levels. All four patients had a moderately to markedly increased urinary glycerol concentration, which was readily detected by GC-MS (Table 1). Only patient 1 had a urinary glycerol concentration in the range of previously investigated GK-deficient patients, whereas it was lower in the other three patients. The plasma glycerol concentration was 10–40 times above the upper reference limit (<0.28 mmol/l) in patients 1, 2 and 3 but within the reference range in patient 4, who also had the lowest urinary glycerol concentration. A specific enzyme assay and two indirect assays demonstrated GK activity levels ranging from severe deficiency in patient 1, to normal GK activity, normal glycerol oxidation to [14C]CO2 and normal [14C]glycerol carbon incorporation into macromolecules in patient 4 (Table 2). Patient 2, who showed classical manifestations of isolated GKD, had a normal incorporation into macromolecules of [14C] from [14C]glycerol, but both the specific GK assay and the glycerol oxidation analysis revealed a significant GK deficiency (22 and 37% of reference value, respectively). In patient 3, the specific assay and the two indirect assays showed partial GK deficiency (16, 17 and 22%, respectively). Kinetic analysis showed that patient 2 had an elevated Km but most probably a normal Vmax, while patients 1 and 3 had a normal Km but a decreased Vmax, the latter being in the same range as when estimated with the enzymatic assay using a glycerol concentration of 45 μmol/l (Figure 1, Table 2). Patient 4 had normal Km and Vmax.

Substrate saturation assay. GK activity measured at different glycerol concentrations from 1 to 110 μmol/l. Patient 4 (▪) and the control (○) have normal Km and Vmax. Patients 1 (□) and 3 (▵) have normal Km but a reduced Vmax, whereas patient 2 (▴) has an elevated Km but probably a normal Vmax.
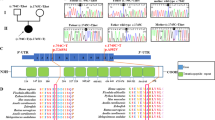
The four patients were tested for GK gene abnormalities. Analysis of the GK exons by electrophoresis on ethidium bromide-stained agarose gels indicated no gross gene abnormalities in any of the patients. Subsequently, the amplified GK exons were sequenced. Patient 1 had a c.1194A>C transversion in exon 14 of the GK gene (Figure 2a), resulting in an E398D amino-acid substitution. Patient 2 showed a c.182T>C transition in exon 3 of the GK gene (Figure 2b), causing an L61P amino-acid substitution. ASOH analysis was used to screen the family members. Both patients were hemizygous and their mothers were heterozygous for the mutations (data not shown). To test whether these are common polymorphisms, we screened 100 alleles from 50 unrelated normal females. All controls had the normal sequence. Patients 3 and 4 did not show any GK gene abnormalities in the exons or in the intron regions in the immediate vicinity of the exons. Patient 3, who was suspected to have additional AHC, did not show any alterations in the DAX1 gene (data not shown).

DNA mutations. (a) Sequence from exon 14 in the GK gene showing the c.1194A>C transversion in DNA from patient 1 and a normal control. (b) Sequence from exon 3 in the GK gene showing the c.182T>C transition in DNA from patient 2 and a normal control. (c) Sequence from the genomic part of intron 3 showing the g.16835G>A mutation in DNA from patient 3 and a normal control (antisense strand).
Furthermore, analysis of mRNA from the Xp 21 GK gene was performed and amplification of the fragment containing exons 1–8 resulted in a fragment with the expected length in patients 1, 2 and 4 (Figure 3a), whereas in patient 3 the amplification resulted in three different fragments 3.1, 3.2 and, 3.3 (Figure 3b). Amplification of exons 8–19 resulted in cDNA fragments with the normal size in all four patients (data not shown). Sequencing of the fragments detected the mutations in patients 1 and 2 earlier found with the exon amplification strategy and no aberration was found in cDNA from patient 4. Sequencing of the three different cDNA fragments from patient 3 (Figure 3b), detected an unknown 212 nt long sequence that spliced to exon 4 in fragments 3.1 and 3.2. The 3.3 fragment had the normal length and the correct cDNA sequence.

RT-PCR analysis of the Xp21 GK gene. (a) Agarose gel showing the amplified fragment from exons 1 to 8. In patient 2 (lane 1–2), patient 4 (lane 6) and patient 1 (lane 7–8), the amplification results in the normal 667 bp fragment as in the control sample (lanes 9–10). In mRNA from patient 3 (lane 3), the analysis results in three fragments. (b) Lanes 3 and 4 show the different cDNA fragments found in mRNA from patient 3. Two longer fragments 3.1 (879 bp) and 3.2 (772 bp) and the normal sized fragment 3.3.
An ≈2.5 kb genomic DNA fragment was amplified using a primer designed from the cryptic exon part and the GKE 3A primer.17 After partial sequencing of the 2.5 kb fragment, a positive identification to a region of the GenBank Accession No. AC005913 was made. This 147 kb BAC clone contains the first exons in the GK gene and the 212 nt sequence acting as a cryptic exon is located in intron 3. Sequencing of this genomic region in DNA from patient 3 revealed a g.16835G>A mutation, in position −2 before the cryptic exon sequence (Figure 2c). The g.16835G>A mutation was not found in 100 X-chromosomes tested.
Complete sequencing of the two longer cDNA transcripts, 3.1 and 3.2, showed that the 212 nt cryptic exon was spliced in between exon 2 or 3 and exon 4 (Figure 4). The g.16835G>A mutation results in three different transcript [r.=,r.153_259delins152+1_152+212, r.259_260ins259+1_259+212]. Translating the cDNA sequence of the two transcripts with the cryptic exon insertion predicts that they consists of premature stop codons and would probably not result in functional proteins.

Effect of the g.16835G>A mutation in patient 3. (a) Schematic representation of the genomic DNA from exons 2 to 4 with part of a cryptic exon sequence from intron 3 indicated. The g.16835G>A mutation in patient 3 (marked in bold) activates a very strong acceptor site. As a result, a 212 nt long part of intron 3 (IVS3) now act as a cryptic exon (the start is marked with capitals). (b) Schematic representation of the three different cDNA fragments from exons 2 to 4 found in patient 3. Fragment 3.1 (879 bp), [r.259_260ins259+1_259+212] with the cryptic exon spliced between exons 3 and 4; fragment 3.2 (772 bp), [r.153_259delins152+1_152+212] lacking exon 3 and with the cryptic exon spliced in between exons 2 and 4; and fragment 3.3 (667 bp) [r=] with a correct splicing of the three exons 2, 3 and 4.
Discussion
GKD is usually suspected in the laboratory from two findings: (a) high glycerol excretion in the urine found by GC-MS in the course of investigation of urinary organic acids (patients 2, 3 and 4), (b) falsely high serum triglyceride concentration as a result of the codetermination of glycerol in the triglyceride method (patient 1).
Further studies of glycerol metabolism are needed when urine and serum glycerol are elevated. Both specific and indirect assays of GK activity are available and are adequate to detect complete GKD (enzyme activity <1–5% of reference).1,6 In the patients reported here, partial GK deficiencies were demonstrated in three of the four cases by specific as well as indirect methods. The three methods give comparable results if the reduced GK activity results from a lower amount of transcript but with a normal enzyme conformation, as in patient 3. Different results were found in patient 2, who was shown to have a mutation affecting the Km for glycerol (Table 2). The specific assay showed a residual GK activity of 22% of control at the glycerol concentration of the standard assay, both indirect assays suggested higher residual activity (37% by glycerol oxidation) or even normal activity (92% by glycerol carbon incorporation). Apparently, under the conditions of the latter indirect assay, the GK activity remains sufficiently high to maintain a normal flow towards synthesis of TCA-precipitable macromolecules. These results emphasize that the apparent level of residual enzyme activity may strongly depend on the conditions of the assay, especially in the case of variants with increased Km or with reduced enzyme stability. Carbon incorporation into protein and glycerol oxidation are assays of glycerol metabolism that require other functioning enzymes than GK, and could therefore also be used in cases of glyceroluria and/or glycerolemia with normal GK activity in attempts to pinpoint other aberrations in glycerol metabolism than GKD.
In patient 4, a moderate glyceroluria – albeit with normal serum glycerol concentration – led to the suspicion of GKD. This diagnosis could be excluded from the measurement of GK activity. None of the indirect methods indicated an aberration in the metabolism of glycerol and thus the cause of the glyceroluria remains unknown.
DNA analysis in patients 1 and 2 showed two novel missense mutations in the GK gene: c.1194A>C (E398D) and c.182T>C (L61P), respectively. The glutamic acid at position 398 is conserved both in prokaryotic and eukaryotic organisms and may therefore be essential for the functioning of the GK protein (Figure 5a). Although the leucine 61 is only conserved in eukaryotes such as rat, mouse and Saccharomyces cerevisiae (Figure 5b), it is located in an α-helix on the outside of the GK subunit that interacts with another subunit to form the functional tetramer.22 As proline residues are prone to interrupt helix formation, the mutation may cause a conformational change that influences the functioning of the GK protein. Also, the decreased affinity for glycerol as shown by the high Km for glycerol in this patient supports the idea of a disturbed tetramer structure. To date, the mutations were not identified in a panel of 100 X-chromosomes of normal controls. It is therefore probable that both missense mutations are disease-causing gene defects.

Amino-acid sequence alignment. Comparison of the (a) glutamic acid at position 398 (E398) and (b) leucine at position 61 (L61) for human GK with those for Rattus norvegicus, Mus musculus and S. cerevisiae (eukaryotes); B. subtilis and E. coli (eubacteria); S. solfataricus and A. fulgidus (archaebacteria) (Comparative sequence analysis, http://coot.embl-heidelberg.de/Alignment/). Amino acids that are identical with those in human GK are shown with asterisks (*). A closed diamond symbol above the sequence (♦) indicates interactions with ADP in E. coli.29
Several cases with a GK activity >5% have been reported earlier. Sargent et al10 have described a C256R substitution in a patient with 12% GK activity. This amino-acid residue is located in a β-sheet close to domains involved in ATP binding,22 and this may result in an enzyme with some residual activity. The insertion of an Alu sequence in intron 4 of the GK gene described by Zhang et al11 does not effect the mRNA as judged from their RT-PCR analysis, but the residual GK activity was 32% of the mean normal control. The mutation in the intron sequence found in our patient 3 results in three different mRNA, one correctly spliced and two transcripts where the open-reading frame is disturbed, with the residual GK activity of 15–20% explained by the small amount of correctly spliced transcript. As discussed before,21 RT-PCR analysis of the GK gene is tricky as there is a high homology between this gene and pseudogenes in the GK gene family. The differently spliced transcripts in our patient 3 were not detected with an RT-PCR using primers described earlier.23 Dipple et al9 have studied six individuals with missense mutations in the GK gene and report GK activities between 5 and 13% of control. They looked into the location of these amino acids in the three-dimensional structure of GK from Escherichia coli. In two of the patients, with ∼12% GK activity, the mutations are located in regions involved in ATP binding. An R405Q substitution is located in the same α-helix as the E398D substitution found in our patient 1.
When exon amplification and sequencing does not detect the mutation one can turn to RT-PCR analysis, as shown in patient 3. A mutation in intron 3 (g.16835G>A) created a cryptic splice acceptor site, resulting in alternative splicing of the transcript with a small amount of correctly spliced GK mRNA that can explain the high residual GK activity (20%) in patient 3. Similar findings were reported in another patient by Sargent et al10 with reduced levels of GK activity.
Splice junction analysis has been used to evaluate the effect of mutations in the intron boundaries.7,21,24 The programs calculate the individual contribution of the nucleotide to the acceptor or donor site. In the cryptic exon sequence including its surroundings, at least four possible acceptor sites exists in the wild type, close to the start of the cryptic exon, but these are probably not functional sites. Three of these have values 6.6, 5.2 and 6.2 bits at locations –1 to –3 relative to the start of the cryptic exon (data not shown). With these very similar values at adjacent locations, they probably compete strongly and are thus nonfunctional in the wild type. The g.16835G>A mutation results in a very strong and functional cryptic splice acceptor site of value 14.7 bits at location –1 relative to this fragment and thus creates a new 212 nt long cryptic exon. The cryptic acceptor site created by the mutation is more than 200 times stronger than the wild-type acceptor sites. The splice sites used for exons 2 and 3 are of moderate strength. The acceptors have values 8.5 and 8.0 bits; the donors, 6.7 and 5.0 bits, respectively. This donor for exon 3 is rather weak. Cases have been reported where upstream exons are skipped when their splice site values are much lower than those of adjacent downstream ones.25 Thus, the extremely strong cryptic acceptor site due to the g.16835G>A mutation could cause both insertion of a cryptic exon and partial skipping of the preceding exon 3, with its weak donor site, as seen in the mRNA analysis.
Three patients presented with clinical symptoms, suggesting isolated or complex (patient 3) GKD. In these patients decreased GK activity and mutations in the GK gene confirmed the diagnosis of GKD. Patients 1 and 2 are typical for the isolated form of GKD from both clinical and laboratory point of view. Although patient 2 had an appreciable GK activity under optimum in vitro conditions, the in vivo flux through the glycerol pathway was apparently too low to prevent the development of symptoms. Patient 1 also has mild mental retardation. Only a few patients with isolated GKD and a mental alteration have been reported before.1,6,9,23 In 38 cases of isolated GKD from 28 families, mental retardation and developmental delay have been reported in five cases.1 We have knowledge of a further 15 cases who are without mental problems (unpublished observation). Our patient 1 also had facial dysmorphism, myopia, strabismus, etc, indicating a developmental defect in addition to GKD. A severely retarded GKD patient reported by Blomquist et al6 also had osteoporoses, multiple fractures and dental problems, which could not be explained by GKD. The clinical data concerning most reported cases are scarce, for example, in degree of mental retardation and developmental delay and follow-up period. In our opinion, the question of mental retardation as a phenotypic trait in isolated GKD9 must be left open. There is, however, the possibility that severe catabolic episode at an early age may cause brain damage. In contrast to the situation in isolated GKD, mental retardation is very common in patients with deletion affecting DAX1, GK and DMD genes possibly by involving the IL1RAPL1 gene. This gene is located distal to the DAX1 gene and part of this gene is deleted in three brothers with a mild Becker-type muscular dystrophy, GKD, AHC and a mild mental retardation.26
No aberration was found in the DAX1 gene in patient 3. Although this patient presented nonspecific clinical features, the clearcut hyperglycerolemia, significant glyceroluria and a deficiency of fetal adrenal zone steroid metabolites led to the suspicion of complex GKD. RNA analysis proved that he had a mutation in the GK gene and thus the isolated form of GKD and not the complex form, as it is defined to be caused by deletion of different loci in the p21 region of the X chromosome. As the g.16835G>A mutation has recently been described in another patient with isolated GKD, normal creatine kinase activity and adrenal function,10 it is unlikely that this mutation could explain the complex clinical picture in our patient. The possibility of another gene defect occurring together with the insertion in the GK gene cannot be excluded. Romero et al27 reported a c.947C>T mutation in the GK gene in a patient with a deletion of exons 13–29 in the DMD gene.
Patient 4 did not have GKD and no abnormalities in the metabolism of glycerol could be detected with the two indirect assays used. This patient showed a delayed development with glyceroluria without hyperglycerolemia similar to five previously reported children with developmental abnormalities.28 However, none of the recognized causes of increased glyceroluria, such as hyperthyroidism, von Gierke's disease, fructose-1,6-biphosphatase deficiency, chronic renal failure and strenuous exercise,28 was present in our patient 4. Also, contamination of urine specimens was ruled out by the consistent presence of increased levels in multiple samples of this patient. The cause of his persistent glyceroluria remains unknown.
We conclude that investigation of patients with glyceroluria should start with the determination of plasma glycerol concentration. Defective glycerol metabolism as a consequence of GKD can be measured in fibroblasts, either with a specific GK assay or indirectly. Indirect GK activity assays by measurement of glycerol carbon incorporation into macromolecules or glycerol oxidation may miss some rare variants with partial GKD. Normal GK activity in indirect test may therefore need further specific enzyme analysis. Genetic analysis may help to explain residual activity and will facilitate the identification of carriers in families.
References
Sjarif DR, Sinke RJ, Duran M et al: Clinical heterogeneity and novel mutations in the glycerol kinase gene in three families with isolated glycerol kinase deficiency. J Med Genet 1998; 35: 650–656.
Hellerud C, Wramner N, Erikson A, Johansson Å, Samuelson G, Lindstedt S : Glycerol kinase deficiency; follow-up during 20 years, genetics, biochemistry, and prognosis. Acta Paediatr, In Press.
McCabe ERB : Disorders of glycerol metabolism; in Scriver CR, Beaudet AL, Sly WS, Valle D (eds): The Metabolic and Molecular Bases of Inherited Disease, 8th edn. New York: McGraw-Hill; 2001, pp 2217–2237.
Scheuerle A, Greenberg F, McCabe ER : Dysmorphic features in patients with complex glycerol kinase deficiency. J Pediatr 1995; 126: 764–767.
Rose CI, Haines DS : Familial hyperglycerolemia. J Clin Invest 1978; 61 (1): 163–170.
Blomquist HK, Dahl N, Gustafsson L et al: Glycerol kinase deficiency in two brothers with and without clinical manifestations. Clin Genet 1996; 50: 375–379.
Hellerud C, Burlina A, Gabelli C, Ellis JR, Nyholm PG, Lindstedt S : Glycerol metabolism and determination of triglycerides - clinical biochemical and molecular findings in six subjects. Clin Chem Lab Med 2003; 41 (1): 46–55.
McCabe ER, Sadava D, Bullen WW, McKelvey HA, Seltzer WK, Rose CI : Human glycerol kinase deficiency: enzyme kinetics and fibroblast hybridization. J Inherit Metab Dis 1982; 5: 177–182.
Dipple KM, Zhang YH, Huang BL et al: Glycerol kinase deficiency: evidence for complexity in a single gene disorder. Hum Genet 2001; 109: 55–62.
Sargent CA, Kidd A, Moore S, Dean J, Besley GT, Affara NA : Five cases of isolated glycerol kinase deficiency, including two families: failure to find genotype:phenotype correlation. J Med Genet 2000; 37: 434–441.
Zhang Y, Dipple KM, Vilain E et al: AluY insertion (IVS4-52ins316alu) in the glycerol kinase gene from an individual with benign glycerol kinase deficiency. Hum Mutat 2000; 15: 316–323.
Sjarif DR, Dorland L, Sperl W et al: Hyperketonaemia in glycerol kinase deficiency. J Inherit Metab Dis 2000; 23: 760–764.
Bonnefont JP, Specola NB, Vassault A et al: The fasting test in paediatrics: application to the diagnosis of pathological hypo- and hyperketotic states. Eur J Pediatr 1990; 150: 80–85.
Malunowicz EM, Mitkowska Z, Bal K et al: Definitive diagnosis of enzymatic deficiencies of steroidogenesis in at-risk newborns and infants by urinary marker analysis using GC/MS-SIM. Horm Res 1997; 48: 243–251.
Bachmann C : Diagnosis of urea cycle disorders. Enzyme 1987; 38: 233–241.
Lowry OH, Rosebrough NJ, Farr AL, Randell RJ : Protein measurement with the Folin phenol reagent. J Biol Chem 1951; 193: 265–275.
Sargent CA, Young C, Marsh S, Ferguson Smith MA, Affara NA : The glycerol kinase gene family: structure of the Xp gene, and related intronless retroposons. Hum Mol Genet 1994; 3: 1317–1324.
Bertina RM, Ploos van Amstel HK, van Wijngaarden A et al: Heerlen polymorphism of protein S, an immunologic polymorphism due to dimorphism of residue 460. Blood 1990; 76: 538–548.
Yanase T, Takayanagi R, Oba K, Nishi Y, Ohe K, Nawata H : New mutations of DAX-1 genes in two Japanese patients with X-linked congenital adrenal hypoplasia and hypogonadotropic hypogonadism. J Clin Endocrinol Metab 1996; 81: 530–535.
den Dunnen JT, Antonarakis SE : Nomenclature for the description of human sequence variations. Hum Genet 2001; 109: 121–124.
Hellerud C, Adamowicz M, Jurkiewicz D et al: Clinical heterogeneity and molecular findings in five polish patients with glycerol kinase deficiency; investigation of two splice site mutations with computerized splice junction analysis and Xp21 gene specific mRNA analysis. Mol Genet Metab 2003; 79: 149–159.
Hurley JH, Faber HR, Worthylake D et al: Structure of the regulatory complex of Escherichia coli IIIGlc with glycerol kinase. Science 1993; 259: 673–677.
Walker AP, Muscatelli F, Stafford AN et al: Mutations and phenotype in isolated glycerol kinase deficiency. Am J Hum Genet 1996; 58: 1205–1211.
Schneider TD : Information content of individual genetic sequences. J Theor Biol 1997; 189: 427–441.
Thompson TE, Rogan PK, Risinger JI, Taylor JA : Splice variants but not mutations of DNA polymerase beta are common in bladder cancer. Cancer Res 2002; 62: 3251–3256.
Jin H, Gardner RJ, Viswesvaraiah R, Muntoni F, Roberts RG : Two novel members of the interleukin-1 receptor gene family, one deleted in Xp22.1–Xp21.3 mental retardation. Eur J Hum Genet 2000; 8: 87–94.
Romero NB, Recan D, Rigal O et al: A point mutation in the glycerol kinase gene associated with a deletion in the dystrophin gene in a familial X-linked muscular dystrophy: non-contiguous gene syndrome involving Becker muscular dystrophy and glycerol kinase loci. Neuromuscul Disord 1997; 7: 499–504.
Kohlschütter A, Seitz HJ, Feldmann B, Lehnert W, Langenbeck U : Glyceroluria in healthy adults, mentally ill adults and children selected for metabolic screening. Clin Chim Acta 1991; 15: 203–207.
Guo W, Worley K, Adams V et al: Genomic scanning for expressed sequences in Xp21 identifies the glycerol kinase gene. Nat Genet 1993; 4: 367–372.
Acknowledgements
DRS, pediatrician, is a staff member of the Department of Child Health, Dr Cipto Mangunkusumo National General Hospital, University of Indonesia – School of Medicine, Jakarta, Indonesia. She is recipient of a grant from the Van Deventer-Maas Foundation, The Netherlands. We acknowledge the technical assistance of Mr PHA van Zon in DNA mutation analysis, Mr VH Garritsen for [14C]glycerol incorporation studies, Dr Isabelle Vernhet (Department of Medical Genetics and Metabolic Disease, CHU Pellegrin, Bordeaux, France) and Dr Wolfgang Getzner (Zentrallabor, Universitätskliniken Innsbruck, Austria) for glycerol analysis in the urine of patients 1 and 2, respectively, and Dr James R Ellis (Division of Bioengineering and Physical Sciences, Office of Research Services, National Institute of Health, Bethesda, USA) for performing the splice site analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sjarif, D., Hellerud, C., Amstel, J. et al. Glycerol kinase deficiency: residual activity explained by reduced transcription and enzyme conformation. Eur J Hum Genet 12, 424–432 (2004). https://doi.org/10.1038/sj.ejhg.5201172
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201172
Keywords
This article is cited by
-
A rare co-occurrence of duchenne muscular dystrophy, congenital adrenal hypoplasia and glycerol kinase deficiency due to Xp21 contiguous gene deletion syndrome: case report
BMC Endocrine Disorders (2021)
-
Reflection of treatment proficiency of hydroxyurea treated β-thalassemia serum samples through nuclear magnetic resonance based metabonomics
Scientific Reports (2019)
-
β-Thalassemia Patients Revealed a Significant Change of Untargeted Metabolites in Comparison to Healthy Individuals
Scientific Reports (2017)
