
Abstract
Autosomal recessive progressive external ophthalmoplegia (PEO) is one clinical disorder associated with multiple mitochondrial DNA deletions and can be caused by missense mutations in POLG, the gene encoding the mitochondrial DNA polymerase gamma. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is another autosomal recessive disorder associated with PEO and multiple deletions of mitochondrial DNA in skeletal muscle. In several patients this disorder is caused by loss of function mutations in the gene encoding thymidine phosphorylase (TP). We report a recessive family with features of MNGIE but no leukoencephalopathy in which two patients carry three missense mutations in POLG, of which two are novel mutations (N846S and P587L). The third mutation was previously reported as a recessive POLG mutation (T251I). This finding indicates the need for POLG sequencing in patients with features of MNGIE without TP mutations.
Similar content being viewed by others


Main
Progressive external ophthalmoplegia (PEO) is a common sign in mitochondrial disorders and often co-occurs with other variable disease features. One form of PEO is associated with multiple deletions of mitochondrial DNA (mtDNA) in skeletal muscle.1 These mtDNA deletions are secondary to an inherited disorder of mtDNA maintenance caused by a defect in a nuclear gene. In recent years, defects in different nuclear genes have been identified that predispose to both dominant and recessive forms of PEO associated with multiple mtDNA deletions. Dominant PEO can be caused by mutations in the genes encoding the heart/skeletal muscle isoform of the adenine nucleotide translocator (ANT1),2 the novel mitochondrial protein Twinkle (C10orf2)3 and the mtDNA polymerase gamma (POLG).4 Autosomal recessive PEO can be caused by mutations in POLG.4,5,6 So far mutations in the gene encoding thymidine phosphorylase (TP) have been exclusively found in patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE).7 MNGIE patients typically have severe gastrointestinal manifestations including recurrent vomiting and intestinal pseudo-obstruction, peripheral neuropathy, ophthalmoplegia, ptosis and cachexia. Various laboratory findings are reported, including leukoencephalopathy on brain MRI.8 Here, we report the finding of recessive missense mutations in POLG in two sisters with a clinical phenotype undistinguishable from MNGIE.
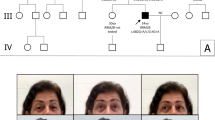
The detailed clinical and laboratory features were described elsewhere.9 Briefly, at age 15 years both patients had onset of gastrointestinal symptoms including chronic pseudo-obstruction. On examination, they had PEO, axonal sensory ataxic neuropathy, muscle weakness and cachexia. Brain MRI was normal. Muscle biopsy demonstrated few ragged red fibers, cytochrome c oxidase negative fibers, decreased enzymatic activities of respiratory chain complexes I and IV, depletion of mtDNA and multiple mtDNA deletions. Direct sequencing revealed homozygosity for a common polymorphism in TP but no pathogenic mutations and enzymatic TP activity in leukocytes was normal.9 The pedigree of the recessive family is shown in Figure 1a.

(a) Segregation analysis of POLG mutations. Squares represent males, circles represent females, filled symbols represent affected individuals and slashed symbols represent deceased individuals. Alleles/haplotypes between brackets were reconstructed from the pedigree data. (b) Position of mutant residues in POLG and amino-acid sequence alignment, showing evolutionary conservation of altered residues.
In both the patients, pathogenic mutations in TP had been excluded previously.9 The fact that axonal predominantly sensory peripheral neuropathy is common in recessive PEO patients with POLG mutations,6 prompted us to analyze POLG. Sequencing of all coding POLG exons using flanking intronic primers4 in the proband (II.4) revealed the presence of three single-base changes. One mutation in exon 3 (c.752C→T), predicting the amino-acid substitution T251I, has already been reported in four Italian compound heterozygote recessive PEO patients of three unrelated families, while absent in Italian control individuals.5 The other two mutations, in exons 10 (c.1760C→T) and 16 (c.2591A→T), are novel and predict P587L and N864S substitutions, respectively. Segregation of the mutations within the family is depicted in Figure 1a. DNA from the deceased affected sister (II.1) had to be recovered from paraffin-embedded tissue and we were only able to demonstrate the presence of T251I in this patient. The occurrence of N864S in her unaffected son (III.1, aged 24 years), however, demonstrated that she is an obligate carrier of this mutation. The unaffected grandmother (I.2) and grandson (III.1) are heterozygote carriers of N864S, indicating that N864S is in trans with the other two mutations, T251I and P587L. The cis configuration of T251I and P587L is confirmed by their heterozygous appearance in the unaffected granddaughter (III.2, aged 12 years). The segregation in the family is consistent with the recessive nature of the three POLG mutations with the two patients being compound heterozygotes. All three mutations (T251I, P587L and N864S) were absent in 280 control chromosomes (90 Belgian and 50 Danish healthy control individuals). Belgian controls were examined using the pyrosequencing technique,10 whereas Danish controls were tested by restriction enzyme digestion (T251I destroys a BsrI site, P587L creates an XmnI site and N864S creates a Bsp1286I site). The three mutations apparently have a low frequency in the population examined (<0.36%) and thus are rare polymorphisms. All three mutations in this family alter evolutionary conserved residues (Figure 1b). Since there are currently no methods available for studying the functional effects of POLG mutations in vivo, we cannot reach a conclusion about the pathogenic nature of the three POLG mutations. Also, we cannot exclude that either all three POLG mutations or one of the two possible trans combinations contribute to the phenotype in the patients.
Our finding expands the clinical spectrum of recessive POLG mutations since severe gastrointestinal features caused by dysmotility and dilation have not been encountered previously in recessive PEO patients with POLG mutations 5,6 while they are prominent manifestations of MNGIE caused by TP mutations.8,11 The more severe phenotype in the present family versus previously described recessive PEO patients with POLG mutations might result from a greater functional impairment of POLG induced by the three mutations together. Also, depending on the location of the mutation within POLG a more or less severe effect on POLG functioning may result. In this respect, it is worth mentioning that N864S involves a highly conserved sequence block close to the POLG polymerase motif A, and that T251I is located between the POLG exonuclease motifs I and II (Figure 1b). Data from the current literature show that the clinical phenotype of recessive PEO patients with POLG mutations is quite heterogeneous. Common features include PEO and axonal sensory neuropathy but associated features are quite variable, including sensory ataxia, cardiomyopathy, retinopathy, sensorineural hearing loss, thalamic lesions, muscle dystrophy, facial diplegia, dysarthria and dysphagia.5,6 This protean presentation of recessive POLG mutations contrasts with the apparently homogeneous clinical picture of MNGIE, which is caused by mutations in TP.8 Only the absence of leukoencephalopathy on brain MRI could distinguish our patients from those with TP mutations. However, in earlier descriptive studies on MNGIE, leukoencephalopathy was not an inclusion criterion.11 On the other hand, leukoencephalopathy has not yet been encountered in patients with POLG mutations,5,6,12 and it has been suggested that in MNGIE patients leukoencephalopathy could be related to the role that TP plays in glial cell proliferation or cortical neuron trophism, which is unrelated to the effect of TP on nucleotide metabolism.7 If further studies on TP and POLG in patients with recessive PEO or MNGIE characterized by multiple mtDNA deletions would confirm these data, brain MRI could be used as a noninvasive test to differentiate patients with TP mutations from those with POLG mutations.
References
Zeviani M, Servidei S, Gellera C, Bertini E, DiMauro S, DiDonato S: An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature 1989; 339: 309–311.
Kaukonen J, Juselius JK, Tiranti V et al: Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 2000; 289: 782–785.
Spelbrink JN, Li FY, Tiranti V et al: Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle a, phage T7 gene 4-like protein localized in mitochondria. Nat Genet 2001; 28: 223–231.
Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C: Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet 2001; 28: 211–212.
Lamantea E, Tiranti V, Bordoni A et al: Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Ann Neurol 2002; 52: 211–219.
Van Goethem G, Martin JJ, Dermaut B et al: Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul Disord 2002; 13: 133–142.
Nishino I, Spinazzola A, Hirano M: Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 1999; 283: 689–692.
Nishino I, Spinazzola A, Papadimitriou A et al: Mitochondrial neurogastrointestinal encephalomyopathy: an autosomal recessive disorder due to thymidine phosphorylase mutations. Ann Neurol 2000; 47: 792–800.
Vissing J, Ravn K, Danielsen ER et al: Multiple mtDNA deletions with features of MNGIE. Neurology 2002; 59: 926–929.
Alderborn A, Kristofferson A, Hammerling U: Determination of single-nucleotide polymorphisms by real-time pyrophosphate DNA sequencing. Genome Res 2000; 10: 1249–1258.
Hirano M, Silvestri G, Blake DM et al: Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology 1994; 44: 721–727.
Van Goethem G, Martin JJ, Lofgren A et al: Unusual presentation and clinical variability in Belgian pedigrees with progressive external ophthalmoplegia and multiple deletions of mitochondrial DNA. Eur J Neurol 1997; 4: 476–484.
Acknowledgements
This work was in part supported by a grant from the Danish National Research Foundation (504-14). BD is a doctoral fellow of the Fund for Scientific Research-Flanders (FWO-F).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Van Goethem, G., Schwartz, M., Löfgren, A. et al. Novel POLG mutations in progressive external ophthalmoplegia mimicking mitochondrial neurogastrointestinal encephalomyopathy. Eur J Hum Genet 11, 547–549 (2003). https://doi.org/10.1038/sj.ejhg.5201002
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201002
Keywords
This article is cited by
-
Pharmacovariome scanning using whole pharmacogene resequencing coupled with deep computational analysis and machine learning for clinical pharmacogenomics
Human Genomics (2023)
-
Role of Mutations of Mitochondrial Aminoacyl-tRNA Synthetases Genes on Epileptogenesis
Molecular Neurobiology (2023)
-
The enteric nervous system in gastrointestinal disease etiology
Cellular and Molecular Life Sciences (2021)
-
Mutations in MTHFR and POLG impaired activity of the mitochondrial respiratory chain in 46-year-old twins with spastic paraparesis
Journal of Human Genetics (2020)
-
Infectious stress triggers a POLG-related mitochondrial disease
neurogenetics (2020)
