
Abstract
Rhabdoid tumour predisposition syndrome (RTPS) is a rare syndrome caused by inheritance of a mutated INI1 gene for which only two multigeneration families have been reported. To further characterise the genotype and phenotype of RTPS, we present a third family in which at least three cousins developed an atypical teratoid/rhabdoid tumour (AT/RT) at a young age. Two of these patients showed unusual long survival, and one of these developed an intracranial meningioma and a myoepithelioma of the lip in adulthood. Mutation analysis of INI1 revealed a germline G>A mutation in the donor splice site of exon 4 (c.500+1G>A) in the patients and in their unaffected fathers. This mutation prevents normal splicing and concomitantly generates a stop codon, resulting in nonsense-mediated mRNA decay. Biallelic inactivation of INI1 in the tumours, except for the meningioma, was confirmed by absence of nuclear INI1-protein staining. The myoepithelioma of one of the patients carried an identical somatic rearrangement in the NF2 gene as the AT/RT, indicating that both tumours originated from a common precursor cell. In conclusion, this study demonstrates for the first time transmission of a germline INI1-mutation in a RTPS family via nonpenetrant males, long-term survival of two members of this family with an AT/RT, and involvement of INI1 in the pathogenesis of myoepithelioma.
Similar content being viewed by others

Main
High-grade malignant brain tumours (MBTs) of childhood include medulloblastoma, primitive neuro-ectodermal tumour, anaplastic astrocytoma, glioblastoma, anaplastic ependymoma, atypical teratoid/rhabdoid tumour (AT/RT), and choroid plexus carcinoma (CPC) (Kalifa and Grill, 2005). Although most of these tumours are sporadic, familial cases have been described in which inherited germline mutations in tumour suppressor genes predispose to their development. Germline mutations in PTCH and TP53 are known to be involved in the familial occurrence of medulloblastoma in childhood in Gorlin syndrome (Johnson et al, 1996) and of various types of brain tumours in Li-Fraumeni syndrome (Frebourg et al, 1995). The familial occurrence of ependymomas has been well documented, but the predisposing gene remains to be identified (Dimopoulos et al, 2006). Familial cases of AT/RT or CPC are very rare. Until now, two families have been described in which the development of these malignant childhood brain tumours correlated with the inheritance in two or more generations of a mutation in the INI1/hSNF5/BAF47/SMARCB1 gene on chromosome 22 (Taylor et al, 2000; Janson et al, 2006). This tumour suppressor gene encodes INI1, which is a core subunit of the chromatin remodelling SWI/SNF complex (Wang et al, 1996; Phelan et al, 1999). In one family, an 18-month-old girl presented with a cerebellar rhabdoid tumour with an INI1 exon 7 donor-splice site mutation, which she inherited from her unaffected carrier mother. A brother of the latter died at the age of 2 years of a tumour diagnosed as CPC with the same mutation (Taylor et al, 2000). In the other family, two half-brothers each developed an AT/RT at a young age and inherited an INI1 insertion mutation in exon 4 from their unaffected carrier mother. Their maternal half-uncle, who was diagnosed with a medulloblastoma and a renal rhabdoid tumour, died at 2 years of age, suggesting that he inherited the same mutation, although this could not be studied (Janson et al, 2006).
Here, we present an extended molecular analysis of a previously reported family in which four cousins were affected with an MBT at young age (Hulsebos et al, 1999). These tumours were originally classified as anaplastic ependymomas (Nijssen et al, 1994). However, because of the previously documented involvement of chromosome 22 in the inheritance of the predisposition to these tumours (Hulsebos et al, 1999) and the possibility of misclassification, we investigated whether these anaplastic ependymomas were in fact AT/RTs. This proved to be the case, as we concluded from re-evaluation of their histopathology and from mutation and expression analysis of the INI1 gene in three of these tumours. In addition, one of the patients developed a meningioma and a myoepithelioma in adulthood.
Patients and methods
Patient material
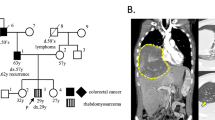
Written informed consent was obtained from patient III-1 (for pedigree see Figure 1). Each of the four cousins developed an MBT at a young age (<5 years). After removal of the tumour, patient III-1 received adjuvant chemotherapy (methotrexate, vincristine, and prednisolone) followed by craniospinal radiotherapy (3300 cGy in 22 doses) with a boost of 2100 cGy on the location of the tumour. He developed an intracranial meningioma and a myoepithelioma of the lip at, respectively, 25 and 26 years of age. Patient III-4 developed a recurrent brain tumour almost 2 years after removal of the primary brain tumour and treatment with chemotherapy (vincristine, procarbazine, and methotrexate). The sister of the paternal grandfather (I-1) of the four cousins died of an uncharacterised brain tumour at the age of 2 years. The clinical findings for the patients are summarised in Table 1.

Haplotyping of family members and LOH analysis of tumours using microsatellite markers from chromosome 22. Patients with the germline INI-mutation are represented by black symbols and proven nonexpressing carriers of this mutation by dotted symbols. Haplotypes were constructed assuming minimal numbers of recombinations. Haplotypes for the grandfather (between brackets) were inferred. N: noninferred marker allele. MBT-1, MBT-3, and MBT-4(R) are the marker alleles retained in the MBT of patients III-1, III-3, and III-4 (R, recurrent tumour), respectively. M-1 and My-1 represent marker alleles retained in meningioma and myoepithelioma of patient III-1, respectively.
Freshly frozen tumour tissue was only available from the meningioma of patient III-1 Formalin-fixed and paraffin-embedded tumour tissue was available from the other tumours.
DNA and RNA samples
Tumour DNA was extracted using commercially available kits (QIAGEN, Venlo, The Netherlands). Extraction of constitutional DNA from blood leukocytes was performed as described previously (Hulsebos et al, 1999).
Microsatellite analysis
Microsatellite markers used for haplotype analysis of family members and loss of heterozygosity (LOH) analysis in tumours were, ordered from 22cen to 22qter, D22S420, D22S427, D22S941, D22S311, D22S446, D22S539, D22S686, D22S303, D22S257, D22S345, D22S156, RH13801, D22S925, D22S926, D22S419, D22S421, D22S1164, D22S429, D22S1148, D22S310, D22S1167, D22S1163, D22S1150, nf2C3.1, D22S929, nf2CAV, D22S1176, D22S280, D22S284, D22S419, D22S1165, D22S1171, D22S274, D22S1169, D22S1145. Because of substantial degradation of template DNA, reliable determination of the allele status of markers generating polymerase chain reaction (PCR) products longer than 150 bp could not be performed for the MBTs of III-1 and III-3 and for the recurrent tumour of III-4. Primer sequences and amplification conditions were taken from the Genome Database at http://www.gdb.org/ or from Legoix et al (1999). Polymerase chain reaction reactions were performed essentially as described before (Bijlsma et al, 1995). 32P-dCTP-labelled PCR products were separated on 6% acrylamide denaturing gels and visualised with autoradiography.
Mutation analysis
The INI1 and NF2 genes were sequenced by using genomic DNA as substrate for amplification by PCR. Primer sequences for mutation analysis of the 9 INI1 exons and the 17 NF2 exons have been given previously (Hulsebos et al, 2007). In degraded DNA samples, the exon 4–intron 4 junction region of INI1 was sequenced using forward primer 5′-catgctccacaaccatcaac-3′ and reverse primer 5′-aactgaaacgtgctggagaac-3′, generating a PCR-product of 131 bp.
Denaturing high-performance liquid chromatography
The exon 4–intron 4 junction region of INI1 was amplified with forward primer 5′-ggatcaggtcctatactgac-3′ and reverse primer 5′-aactgaaacgtgctggagaac-3′, generating a product of 248 bp. Polymerase chain reaction products were analysed on an Agilent 1100 system (Agilent, Amstelveen, The Netherlands) equipped with a Helix DNA column (Varian, Middelburg, The Netherlands). Denaturing high-performance liquid chromatography (DHPLC) running conditions are available upon request.
Multiple ligation-dependent probe amplification
Multiple ligation-dependent probe amplification (MLPA) analysis of the NF2 gene was performed using the SALSA P044 NF2 MLPA Kit according to the instructions of the manufacturer (MRC-Holland, Amsterdam, The Netherlands).
Immunohistochemical analysis
Immunohistochemical analysis of the INI1 protein was performed using a BAF47/INI1 antibody (BD Transduction Laboratories, Franklin Lakes, NJ, USA) on formalin-fixed, paraffin-embedded tumour tissue as described previously (Hulsebos et al, 2007).
Results
Mutation analysis of INI1
Sequencing of the 9 INI1-exons and their flanking sequences in the constitutional blood DNA of patient III-1 revealed a mutation in the donor splice site of exon 4 (Figure 2). This heterozygous G>A mutation alters the conserved GT sequence at the beginning of intron 4 (c.500+1G>A of the mRNA (GenBank accession number U04847)), enabling read-through of the transcript at this position. Concomitantly, the G>A mutation results in the generation of an in-frame stopcodon (UGG → UGA, p.W167X) in the read-through transcript. This mutation was also present in the constitutional DNA of patient III-4 and of the unaffected fathers II-2 and II-3. As exemplified in Figure 2 for tumour MBT-1, the malignant brain tumours of patients III-1(MBT-1), III-3 (MBT-3), and III-4 (MBT-4, MBT-4R) and the myoepithelioma of patient III-1 (My-1), but not his meningioma (M-1), showed loss of the wild-type G-allele and retention of the mutant A-allele. Mothers II-1 and II-4 and also grandmother I-2 had only the wild-type sequence in their constitutional DNA (data not shown). The sequencing data for INI1 are summarised in Figure 1. To exclude that the G>A donor splice site mutation represents a polymorphism, we screened the constitutional DNA of 132 normal individuals (264 chromosomes 22) by DHPLC analysis of PCR products containing the exon 4–intron 4 junction region in INI1 but found no mutation-specific profile.

Sequence analysis of the exon 4–intron 4 boundary of INI1. Constitutional heterozygosity for the G>A mutation in blood DNA of patient III-1 (B) but not in normal DNA (A) and retention of the mutant A-allele in MBT DNA of patient III-1 (MBT-1) (C).
Immunohistochemical analysis of INI1 protein expression
In about 3% of cases, the normal GT donor splice site signal at the exon 4–intron 4 junction is ignored during transcription of INI1 and splicing occurs at a cryptic splice site (GC), which is 54 bp distal to the normal splice site (GenBank accession number AK024025) (Favre et al, 2003). Mutation of the GT donor splice site may increase the rate of this alternative splicing. However, the concomitant generation of a stop-codon confers the alternatively spliced transcript in an excellent target for nonsense-mediated decay (Nagy and Maquat, 1998), resulting in absence of INI1 protein encoded by the mutated allele. Alternatively, the mutation may result in skipping of exon 4, generating a transcript that is 138 bases shorter than the wild-type transcript and an INI protein with an in-frame deletion of 46 amino-acid residues. To determine the presence of INI1 protein in the tumours, we performed immunohistochemistry with an INI1-antibody on available histological sections of tumours. The antibody is directed against the C-terminal part of the protein (amino-acid residues 257–359), which remains intact after the eventual in-frame deletion of 46 amino-acid residues (nrs122–167) resulting from exon-4 skipping. As shown in Figure 3, nuclear staining was absent in tumour cells of the recurrence of the brain tumour of patient III-4 (MBT-4R) and in the myoepithelioma of patient III-1 (My-1), but abundantly present in all cells of the meningioma (M-1) of patient III-1. As our sequencing data demonstrated that the normal INI1 gene was lost in the recurrent brain tumour and in the myoepithelioma, but remained in the meningioma, this staining pattern is consistent with the notion that the mutant allele does not encode INI1 protein or shortened INI1 protein as the consequence of exon-4-skipping.

Immunohistochemical INI1-staining of the recurrent MBT of patient III-4 (MBT-4R) (A) and the meningioma (M-1) (B) and myoepithelioma (My-1) (C) of patient III-1. Note absence of nuclear staining of tumour cells in MBT-4R and myoepithelioma. In contrast, the nuclei of endothelial cells of the blood vessels in both tumours and the nuclei of all cells in the meningioma show unequivocal staining. Original magnification × 200.
Microsatellite analysis with chromosome 22 markers
Our sequencing data indicated that the normal INI1 gene was lost in all analysed tumours, except for the meningioma. To determine the mechanism by which these losses occurred, we extended previously reported haplotype and LOH analyses (Hulsebos et al, 1999). The key genetic events are summarised in Figure 1. The LOH analyses indicate that loss of the normal INI1 gene is caused by the deletion of a complete copy of chromosome 22 in the MBTs, but by interstitial deletion in the myoepithelioma. Furthermore, we noted the aberrant retention of allele 1 of marker D22S929 in the brain tumour as well as in the myoepithelioma of patient III-1 (see Figure 1, Figure 4). Flanking markers nf2C3.1 and nf2CAV, however, showed normal retention of the paternal alleles in the myoepithelioma of this patient. As the three markers are located within the NF2 gene (Legoix et al, 1999), we investigated whether this gene was affected by the rearrangement. Sequencing of the 17 exons of NF2 in constitutional, meningioma, and myoepithelioma DNA of patient III-1 revealed no mutations. Multiple ligation-dependent probe amplification analysis of the NF2 gene demonstrated no additional copy number aberrations in blood or tumours of this patient (data not shown).

Microsatellite analysis with intragenic NF2 markers nf2C3.1 (A) D22S929 (B) and nf2CAV (C) of constitutional DNAs of patient III-1, mother II-1, and father II-2, and of tumour DNAs of patient III-1. Positions of alleles are indicated on the left. MBT-1, M-1, and My-1 represent retained alleles in the MBT, meningioma, and myoepithelioma of patient III-1, respectively.
Discussion
Inheritance of the INI1-mutation
We have shown by sequence analysis that the unaffected fathers of the patients were carriers of the germline mutation. As the paternal grandfather's sister died of a brain tumour at 2 years of age and his sons transmitted the mutation, it is most likely that the grandfather is also an unaffected carrier and that his sister developed the brain tumour because of inheritance of the INI1-mutation. This is also supported by the deduced haplotypes of the grandfather's copies of chromosome 22. This study thus documents the inheritance of a germline INI1-mutation that predisposes to the development of MBTs at young age in three subsequent generations of a family. Inheritance of an INI1-mutation via asymptomatic carriers has been reported before in two other families with childhood brain tumours (Taylor et al, 2000; Janson et al, 2006). Although the analyses of the latter families suggested that the asymptomatic transmission of the INI1-mutation exclusively occurred via female carriers, it is clear from our family that the transmission can as well take place via healthy male carriers.
Malignant brain tumours are AT/RTs
Originally, based on their (immuno)histological, ultrastructural, and clinical characteristics, the MBTs of the patients in our family were classified as anaplastic ependymomas (Nijssen et al, 1994). Here, we demonstrate the biallelic inactivation by mutation and deletion of INI1 in the MBTs of patients III-1, III-3, and III-4 and the absence of INI1-protein expression in the recurrence of patient III-4 (Figure 2, Figure 3, respectively). INI1 mutations have not been described in ependymomas (Sevenet et al, 1999a; Kraus et al, 2001; Weber et al, 2001), whereas biallelic inactivation of INI1 and loss of INI1 protein expression are characteristic features of AT/RTs (Versteege et al, 1998; Judkins et al, 2004; Biegel, 2006). Moreover, re-evaluation revealed rhabdoid cells in histological sections of the brain tumours (Table 1). Our data thus show that the brain tumours in at least three of the four cousins are in fact AT/RTs. Especially, as correct histopathological classification of paediatric brain tumours can be challenging (Judkins et al, 2005; Haberler et al, 2006), our present findings suggest that testing for the mutational status of INI1 is warranted to exclude rhabdoid tumour predisposition syndrome (RTPS) in previous (Dimopoulos et al, 2006) and future reports on familial ependymoma.
INI1 inactivation in the tumours of patient III-1
Patient III-1 developed an intracranial meningioma and a myoepithelioma of the upper lip, respectively, 21 and 22 years after operation and chemo- and radiotherapy for the AT/RT.
The meningioma had no mutation in the NF2 gene and displayed a complex karyotype, as evidenced by single-nucleotide polymorphism analysis, including extensive losses on chromosome arm 1p, but no loss of chromosome 22 (data not shown). These molecular and also the clinical characteristics of the meningioma strongly suggest that the development of this tumour is radiation-induced and not part of the RTPS. The biallelic inactivation of INI1 and loss of INI1 protein-expression in the myoepithelioma is remarkable. This benign tumour type is generally characterised by a paucity of genetic alterations and involvement of chromosome 22 or INI1 in the development of this tumour has never been reported before (Hungermann et al, 2002).
NF2 rearrangement in the tumours of patient III-1
Another remarkable finding is the aberrant retention of the maternal allele 1 of marker D22S929 in NF2 on the paternal copy of chromosome 22 that remained in the AT/RT as well as in the myoepithelioma of patient III-1 (Figure 1, Figure 4). This might be explained by assuming a somatic double recombination (or gene conversion) event in a common precursor cell for both tumours, substituting allele 2 for allele 1 on the paternally derived chromosome 22. Although we were unable to demonstrate that the recombination event directly affected the coding regions of NF2, we nevertheless conclude that both tumours must have a common origin. Several patients with a constitutional INI1 mutation and with two tumours at different sites have been reported (Sevenet et al, 1999b; Savla et al, 2000; Kusafuka et al, 2004; Biegel, 2006; Giunti et al, 2006; Meyers et al, 2006). It will be of interest to determine whether the tumours of these patients, like those of patient III-1, are primary tumours, share somatic changes in the genome, and originate from a common precursor cell as well.
In conclusion, we have documented the variable expression and incomplete penetrance of an INI1 germline mutation in a third multigeneration family with the RTPS (Wesseling et al, 2007). Inheritance of the mutation resulted in the development early in life of an AT/RT in at least three cousins. Two of these patients survived this tumour for over 15 years and one of these developed a (probably radiation induced) meningioma and a myoepithelioma in adulthood. We recently showed (Hulsebos et al, 2007) that INI1 is a predisposing gene in familial schwannomatosis, a disorder in which patients develop multiple schwannomas later in life and that is also characterised by variable expression and incomplete penetrance (MacCollin et al, 2003). It has been hypothesised that in patients with a germline INI1 mutation, a developmental window exists at young age in which most rhabdoid tumours occur (Janson et al, 2006). Carriers of the INI1 mutation that do not develop such a tumour at young age might be at increased risk of developing other INI1-related, but not necessarily malignant tumours later in life.
Given the small number of splice site mutations in malignant rhabdoid tumours reported to date (Biegel, 2006), it is interesting to note that our family and one other multigeneration family with RTPS (Taylor et al, 2000) demonstrate an INI1 mutation at a splice site, raising the possibility that carriers of such a mutation may be less likely to be affected. In case of our family, there is the additional possibility that in the brain of the unaffected carriers alternative splicing of the INI1-mutant allele transcript, leading to absence of INI1 protein in the brain tumours of the patients, does not occur. Instead, skipping of exon 4, resulting in the in-frame deletion of 46 amino-acid residues in the INI1 protein, may occur. This truncated protein may still be (partly) functional, at least not contributing to the development of brain tumours in these carriers. Finally, whereas long-term survival in children with AT/RT is extremely rare (Tekautz et al, 2005; Squire et al, 2007), two of the four cousins we described survive for more than 15 years now. More multigeneration families with the RTPS need to be investigated to further elucidate genotype–phenotype correlations in this syndrome.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Biegel JA (2006) Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus 20: 1–7
Bijlsma EK, Voesten AM, Bijleveld EH, Troost D, Westerveld A, Merel P, Thomas G, Hulsebos TJ (1995) Molecular analysis of genetic changes in ependymomas. Genes Chromosomes Cancer 13: 272–277
Dimopoulos VG, Fountas KN, Robinson JS (2006) Familial intracranial ependymomas. Report of three cases in a family and review of the literature. Neurosurg Focus 20: 1–5
Favre M, Butticaz C, Stevenson B, Jongeneel CV, Telenti A (2003) High frequency of alternative splicing of human genes participating in the HIV-1 life cycle: a model using TSG101, betaTrCP, PPIA, INI1, NAF1, and PML. J Acquir Immune Defic Syndr 34: 127–133
Frebourg T, Barbier N, Yan YX, Garber JE, Dreyfus M, Fraumeni Jr J, Li FP, Friend SH (1995) Germ-line p53 mutations in 15 families with Li-Fraumeni syndrome. Am J Hum Genet 56: 608–615
Giunti L, Bernini G, Forni M, Tucci F, Wheeler E, Sardi I (2006) Clonality analysis of pediatric multiple tumors: two case reports and laboratory investigation. J Pediatr Hematol Oncol 28: 241–248
Haberler C, Laggner U, Slavc I, Czech T, Ambros IM, Ambros PF, Budka H, Hainfellner JA (2006) Immunohistochemical analysis of INI1 protein in malignant pediatric CNS tumors: lack of INI1 in atypical teratoid/rhabdoid tumors and in a fraction of primitive neuroectodermal tumors without rhabdoid phenotype. Am J Surg Pathol 30: 1462–1468
Hulsebos TJ, Oskam NT, Bijleveld EH, Westerveld A, Hermsen MA, van den Ouweland AM, Hamel BC, Tijssen CC (1999) Evidence for an ependymoma tumour suppressor gene in chromosome region 22pter-22q11.2. Br J Cancer 81: 1150–1154
Hulsebos TJ, Plomp AS, Wolterman RA, Robanus-Maandag EC, Baas F, Wesseling P (2007) Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet 80: 805–810
Hungermann D, Roeser K, Buerger H, Jakel T, Loning T, Herbst H (2002) Relative paucity of gross genetic alterations in myoepitheliomas and myoepithelial carcinomas of salivary glands. J Pathol 198: 487–494
Janson K, Nedzi LA, David O, Schorin M, Walsh JW, Bhattacharjee M, Pridjian G, Tan L, Judkins AR, Biegel JA (2006) Predisposition to atypical teratoid/rhabdoid tumor due to an inherited INI1 mutation. Pediatr Blood Cancer 47: 279–284
Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein Jr EH, Scott MP (1996) Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272: 1668–1671
Judkins AR, Burger PC, Hamilton RL, Kleinschmidt-DeMasters B, Perry A, Pomeroy SL, Rosenblum MK, Yachnis AT, Zhou H, Rorke LB, Biegel JA (2005) INI1 protein expression distinguishes atypical teratoid/rhabdoid tumor from choroid plexus carcinoma. J Neuropathol Exp Neurol 64: 391–397
Judkins AR, Mauger J, Ht A, Rorke LB, Biegel JA (2004) Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol 28: 644–650
Kalifa C, Grill J (2005) The therapy of infantile malignant brain tumors: current status? J Neurooncol 75: 279–285
Kraus JA, de Millas W, Sorensen N, Herbold C, Schichor C, Tonn JC, Wiestler OD, von Deimling A, Pietsch T (2001) Indications for a tumor suppressor gene at 22q11 involved in the pathogenesis of ependymal tumors and distinct from hSNF5/INI1. Acta Neuropathol (Berl) 102: 69–74
Kusafuka T, Miao J, Yoneda A, Kuroda S, Fukuzawa M (2004) Novel germ-line deletion of SNF5/INI1/SMARCB1 gene in neonate presenting with congenital malignant rhabdoid tumor of kidney and brain primitive neuroectodermal tumor. Genes Chromosomes Cancer 40: 133–139
Legoix P, Legrand MF, Ollagnon E, Lenoir G, Thomas G, Zucman-Rossi J (1999) Characterisation of 16 polymorphic markers in the NF2 gene: application to hemizygosity detection. Hum Mutat 13: 290–293
MacCollin M, Willett C, Heinrich B, Jacoby LB, Acierno Jr JS, Perry A, Louis DN (2003) Familial schwannomatosis: exclusion of the NF2 locus as the germline event. Neurology 60: 1968–1974
Meyers SP, Khademian ZP, Biegel JA, Chuang SH, Korones DN, Zimmerman RA (2006) Primary intracranial atypical teratoid/rhabdoid tumors of infancy and childhood: MRI features and patient outcomes. AJNR Am J Neuroradiol 27: 962–971
Nagy E, Maquat LE (1998) A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 23: 198–199
Nijssen PC, Deprez RH, Tijssen CC, Hagemeijer A, Arnoldus EP, Teepen JL, Holl R, Niermeyer MF (1994) Familial anaplastic ependymoma: evidence of loss of chromosome 22 in tumour cells. J Neurol Neurosurg Psychiatry 57: 1245–1248
Phelan ML, Sif S, Narlikar GJ, Kingston RE (1999) Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol Cell 3: 247–253
Savla J, Chen TT, Schneider NR, Timmons CF, Delattre O, Tomlinson GE (2000) Mutations of the hSNF5/INI1 gene in renal rhabdoid tumors with second primary brain tumors. J Natl Cancer Inst 92: 648–650
Sevenet N, Lellouch-Tubiana A, Schofield D, Hoang-Xuan K, Gessler M, Birnbaum D, Jeanpierre C, Jouvet A, Delattre O (1999a) Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype–phenotype correlations. Hum Mol Genet 8: 2359–2368
Sevenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O (1999b) Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 65: 1342–1348
Squire SE, Chan MD, Marcus KJ (2007) Atypical teratoid/rhabdoid tumor: the controversy behind radiation therapy. J Neurooncol 81: 97–111
Taylor MD, Gokgoz N, Andrulis IL, Mainprize TG, Drake JM, Rutka JT (2000) Familial posterior fossa brain tumors of infancy secondary to germline mutation of the hSNF5 gene. Am J Hum Genet 66: 1403–1406
Tekautz TM, Fuller CE, Blaney S, Fouladi M, Broniscer A, Merchant TE, Krasin M, Dalton J, Hale G, Kun LE, Wallace D, Gilbertson RJ, Gajjar A (2005) Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23: 1491–1499
Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394: 203–206
Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, Crabtree GR (1996) Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev 10: 2117–2130
Weber M, Stockhammer F, Schmitz U, von Deimling A (2001) Mutational analysis of INI1 in sporadic human brain tumors. Acta Neuropathol (Berl) 101: 479–482
Wesseling P, Biegel JA, Eberhart C, Judkins AR (2007) Rhabdoid tumour predisposition syndrome. In: WHO Classification of Tumours of the Central Nervous System, Louis DN et al (eds) Lyon: IARC Press
Acknowledgements
We express our gratitude to Dr PJ Slootweg (Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands) and to Dr R Forsyth (Ghent University Hospital, Ghent, Belgium) for histological advice with regard to the myoepithelioma. We thank A Gemmink (Department of Pathology, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands) for performing the immunohistochemical stainings. This study was supported by the Dutch Cancer Society, grant nos UVA 2001-2561 (TJMH) and KUN 2003-2975 (PW).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Ammerlaan, A., Ararou, A., Houben, M. et al. Long-term survival and transmission of INI1-mutation via nonpenetrant males in a family with rhabdoid tumour predisposition syndrome. Br J Cancer 98, 474–479 (2008). https://doi.org/10.1038/sj.bjc.6604156
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6604156
Keywords
This article is cited by
-
Current recommendations for clinical surveillance and genetic testing in rhabdoid tumor predisposition: a report from the SIOPE Host Genome Working Group
Familial Cancer (2021)
-
Germline variants in SMARCB1 and other members of the BAF chromatin-remodeling complex across human disease entities: a meta-analysis
European Journal of Human Genetics (2018)
-
The molecular pathogenesis of schwannomatosis, a paradigm for the co-involvement of multiple tumour suppressor genes in tumorigenesis
Human Genetics (2017)
-
Atypical teratoid/rhabdoid tumor in the sella turcica of an elderly female with a distinct vascular pattern and genetic alterations
Virchows Archiv (2016)
-
Rhabdoid Variant of Myoepithelial Carcinoma, with EWSR1 Rearrangement: Expanding the Spectrum of EWSR1-Rearranged Myoepithelial Tumors
Head and Neck Pathology (2015)
