
Abstract
Inflammation is a healthy response to infection or danger and should be rapid, specific and terminated once the threat has passed. Inflammatory diseases, where this regulation fails, cause considerable human suffering. Treatments have successfully targeted pro-inflammatory cytokines, such as tumor-necrosis factor (TNF), that directly induce genes encoding inflammatory products. Inflammatory signals, including TNF, may also directly induce caspase-independent cell death (necroptosis), which can also elicit inflammation. Necroptosis was originally defined as being dependent on the kinase RIPK1 but is now known to be dependent on RIPK3 and the pseudo-kinase MLKL. Therefore, RIPK1, RIPK3 and MLKL are potential therapeutic targets. RIPK1 and RIPK3 also directly regulate inflammatory signaling, which complicates interpretation of their function but might alter their therapeutic utility. This Review examines the role of cell death, particularly necroptosis, in inflammation, in the context of recent insights into the roles of the key necroptosis effector molecules RIPK1, RIPK3 and MLKL.
Similar content being viewed by others
Main
Historically, cell death was divided into two forms: the programmed cell death apoptosis, widely considered to prevent inflammation; and unregulated and accidental cell death, necrosis, which was considered to induce inflammation. Now a regulated cellular necrosis called 'necroptosis' has been found to cause the release of cellular contents. Whether necroptosis dependent on the kinase RIPK3 and the pseudo-kinase MLKL is a physiological inducer of inflammation remains contentious. This is partly because the key necroptotic effectors RIPK1 and RIPK3 also directly initiate or regulate inflammatory signaling pathways and there is a dearth of in vivo markers of necroptosis1. As a result, genetic experiments have led the way in tackling these questions1. This Review presents recent insights into cell death and, more specifically, the necroptotic cell-death pathway and its inflammatory role, obtained with newly developed genetic tools. Some of these insights have been gained by attempts to understand why RIPK1-deficient (Ripk1−/−) mice die soon after birth, which had been a mystery for the past 15 years2 (Fig. 1).

A wealth of genetic studies have paved the way to greater understanding of the function of RIPK1, RIPK3 and MLKL in embryonic development and beyond. Key developmental 'checkpoints' in mice have emerged that are heavily under the control of these cell-death regulators, such as those at E10.5 and birth (P0). TNF-TNFR1 signaling and Myd88-dependent pathways have proven important in many of these phenotypes but not universally so, which is probably reflective of the multiple pathways that converge on cell-death signaling and pro-inflammatory cytokine production. P3–P4, post-natal days 3–4; pers. comm., S. Alvarez-Diaz and A. Strasser, personal communication.
Inflammation and damage-associated molecular patterns
Inflammation is characterized by redness, heat, swelling and pain3. These symptoms are driven mainly by cells within the tissue that respond to damage or danger by producing cytokines and chemokines that permeabilize blood vessels and recruit cells of the innate and adaptive immune systems to deal with the insult. Although inflammation helps protect from infection and encourages wound healing, it can be damaging if unregulated, as highlighted by autoimmune and autoinflammatory diseases. The inflammation-inciting agent may be a foreign molecule, frequently associated with a pathogen (a pathogen-associated molecular pattern (PAMP)) or it may be an endogenous but inappropriate molecule (a damage-associated molecular pattern (DAMP)). These DAMPs and PAMPs are detected by usually constitutively expressed receptors, including the NLR, MAVS, RIG-I and AIM2 families of intracellular receptors and the TLR family of extracellular receptors. These signaling receptors enlist similar transcriptional activating factors such as NF-κB, Fos-Jun and IRFs, to activate the production of inflammatory cytokines and chemokines.
The system for detecting and responding to bacterial, viral and parasitic pathogens with the aforementioned receptors is stunningly context sensitive. For example, non-pathogenic organisms often express the same inflammation-inducing molecules as pathogenic ones do, yet mammals co-exist harmoniously with billions of such commensal organisms. Without this context sensitivity, these 'harmless' commensals can trigger a fierce inflammatory response and elicit debilitating inflammation, such as inflammatory bowel disease.
Polly Matzinger proposed context sensitivity could be achieved if the immune system recognized 'danger', rather than pathogens themselves4. But how do cells recognize danger? One obvious alarm to a cell that its tissue residence is endangered is dying or damaged neighbors; thus, the concept of DAMPs was born. However, this raises subsidiary questions. What might a cell death–associated DAMP be? What distinguishes the billions of homeostatic cell deaths that occur in the human body every day from dangerous cell death? The second question is usually answered by the statement that normal homeostatic and developmental cell death occurs via apoptosis, which restricts leaking contents, degrades DNA and generates 'find-me' and 'eat-me' signals to stimulate rapid phagocytic clearance5. However, this appears to contradict another established dogma that apoptosis evolved to limit viral replication and survival6. One way out of this conundrum could be that a viral infection generates additional DAMPs that override the anti-inflammatory nature of apoptosis7. Alternatively, excessive virus-induced apoptosis may overwhelm local phagocytosis and allow these apoptotic cells to undergo secondary 'accidental' necrosis, with release of DAMPs into the tissue8. In support of this idea, failure to clear DNA released from apoptotic cells can lead to autoimmune or autoinflammatory responses9,10. These explanations re-emphasize the proposal that the context is potentially as important as the type of cell death in determining whether it is recognized as dangerous. Finally, during necroptosis, as with accidental necrosis, phagocytes will not be recruited by apoptotic signals and DAMPs will be released extracellularly.
Tumor-necrosis factor and interleukin 1
Tumor-necrosis factor (TNF) and the interleukin 1 (IL-1) family of cytokines are potent inducers of chemokine and cytokine cascades that collectively orchestrate inflammation. Blockade of signaling via TNF and/or IL-1 has proven effective in treating some, but not all, inflammatory diseases11,12. Concentrations of these potent cytokines are tightly regulated, and they are usually present only in very small amounts until induced in response to PAMPs and DAMPs. Furthermore, IL-1β is produced in an inactive precursor ('pro-') form in response to PAMPs or DAMPs and requires a second independent danger signal to cause its processing and cellular release, in a caspase-1-dependent manner13,14, which causes cell death, dubbed 'pyroptosis' because of its association with release of the pyrogen IL-1β. Although IL-1β and IL-1α are released from cells undergoing a pyroptotic death and could therefore also be considered DAMPs, some cells, such as neutrophils do not require lysis to secrete these cytokines15, and kinetic analyses of other cells suggest that both are processed and released before cell death16. It is therefore unclear whether either can be considered a true DAMP in all cases.
TNF powerfully induces inflammatory cytokines, but full appreciation of its inflammatory role requires awareness that TNF can, in certain circumstances, cause cell death and therefore potentially generate DAMPs. This is also true for other members of the TNF super-family, such as FasL and TRAIL, but for simplicity's sake, we will restrict our discussion to TNF and its ubiquitously expressed receptor, TNFR1. The binding of TNF to TNFR1 stimulates the formation of an intracellular complex consisting of recruitment and effector proteins17, which are not the same for all cell types, but we will consider a cell that recruits the adaptors TRADD and RIPK1 to TNFR1 via their respective death domains. TRADD recruits members of the TRAF ('TNFR-associated factor') family and thereby ubiquitin ligases of the cIAP family. Thereafter, cIAPs decorate proteins within this complex with ubiquitin chains that recruit a secondary E3 ligase complex called 'LUBAC', which in turn generates linear ubiquitin chains. Together these ubiquitin chains serve as a platform upon which complexes of the kinases IKK1 and IKK2 and adaptor NEMO (IKKγ), and complexes of the ubiquitin-binding partners TAB2 and TAB3 and the kinase TAK1, are activated. This leads to activation of the transcription factors NF-κB and AP-1 that drive the production of inflammatory cytokines and also the caspase-8 inhibitor cFLIP (Fig. 2). A similar ubiquitin platform–generating cassette has also been described for TLRs that detect PAMPs18,19.

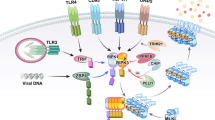
Marina Corral Spence/Nature Publishing Group
Necroptosis can be induced by death receptors including TNFR1 (top left); TLR4, via a TRIF-RIPK1-RIPK3 complex (top right); TLR3, via a TRIF–RIPK1–FADD–cFLIP–caspase-8 complex (middle right); or IFNRs, via a RIPK1-RIPK3 complex (top, middle). These pathways can induce the association of RIPK1 with RIPK3 via RHIM-RHIM domain interactions (light blue) and phosphorylation of RIPK3, which leads to aggregation of phosphorylated RIPK3 (pRIPK3) and phosphorylation of MLKL by RIPK3. Translocation of phosphorylated MLKL (pMLKL) to the cell membrane leads to necroptosis, with release of DAMPs. During infection with murine cytomegalovirus (MCMV), the intracellular receptor DAI, which recognizes viral double-stranded DNA (dsDNA), can directly recruit RIPK3 via a RHIM-RHIM interaction (middle right) and induce phosphorylation and oligomerization of RIPK3, which leads to RIPK1-independent necroptosis. Necroptosis generally occurs only if pro-survival transcriptional and/or apoptotic pathways are compromised. This is exemplified by TNFR1 signaling (apoptotic and transcriptional arms of other pathways not shown here). After it binds TNF, TNFR1 recruits TRADD and RIPK1 to complex I via their respective death domains. TRADD recruits TRAF2 and cIAPs, then cIAPs ubiquitinate components of complex I, which leads to the recruitment of TAK1-TAB1-TAB2, NEMO-IKK1-IKK2 and LUBAC. The LUBAC E3 ligase complex generates linear ubiquitin chains, including chains on RIPK1, which bind additional NEMO. TAK1 phosphorylates the IKK complex, which in turn phosphorylates the NF-κB inhibitory protein IkBa, marking it for ubiquitination followed by proteasomal degradation (not shown here). Translocation of NF-κB to the nucleus then ensues. TNFR1 also induces signaling via AP-1 (not shown here). Transcriptional targets of TNFR1 include the caspase-8 inhibitor cFLIP and the deubiqutinase A20. Deficient complex I activity, such as with loss of cIAPs, can lead to formation of the cytosolic complex II that can induce either apoptosis or necroptosis. cFLIP binds this FADD–RIPK1–caspase-8 complex and inhibits auto-processing of caspase-8 and apoptosis. cFLIP–FADD–RIPK1–caspase-8 simultaneously inhibits necroptosis, probably in part due to its ability to cleave RIPK1. Certain events can promote RIPK3-MLKL–dependent necroptosis, such as depletion of cIAP with concurrent caspase inhibition, deficiency in RIPK1, FADD or caspase-8, or deubiquitination of RIPK1 by the deubiquitinating enzyme CYLD or A20. A20 also has E3 ubiquitin ligase activity and can ubiquitinate RIPK1 via Lys48 and trigger its proteasomal degradation. A20 can also prevent necroptosis by deubiquitinating RIPK3. K63 and K48, Lys63 and Lys48 (ubiquitin linkage); IFN, interferon; LPS, lipopolysaccharide; mtDNA, mitochondrial DNA.
Because pathogens limit inflammation by targeting TNF-TNFR1 and TLR-induced cytokine production, these signaling pathways have evolved to counter such manipulation. If the cIAP-LUBAC–dependent NF-κB response is disrupted, cytokine production is limited, but so is cFLIP production. This may lead to the formation of a secondary caspase-8-dependent, apoptosis-inducing complex20,21 (Fig. 2). In response, some pathogens have evolved to target the cytotoxic activity of TNF by encoding caspase-8 inhibitors22,23. Necroptosis has probably been selected for a host counter-response to this pathogen strategy, because it is enacted when caspases are inhibited and is also favored by conditions that might be induced by pathogens, such as small amounts of ubiquitination of RIPK1 and loss of cIAP or LUBAC activity24,25,26,27. Thus, necroptosis possibly serves as a backup inflammatory response initiated in the context of a pathogen insurgence (Fig. 2).
RIPK1-, RIPK3- and MLKL-induced necroptosis
Until recently, the prevailing model for the role of kinases of the RIP family in necroptosis was that RIPK1 activates RIPK3 (Fig. 2). Direct phosphorylation of RIPK3 by RIPK1 has not been demonstrated; therefore, it is likely that oligomerization of RIPK3 driven by the RHIM domain of RIPK1 and RIPK3 leads to RIPK3 autoactivation28. Consistent with that, other RHIM-containing proteins, such as TRIF and DAI, can promote RIPK3 activation29,30. Activation of RIPK3 leads to the phosphorylation of MLKL and its translocation to the membrane and disruption of the plasma membrane (Fig. 2). Thus, MLKL appears to be a key necroptotic effector, but exactly how it disrupts membranes is unclear, because published reports have provided different and in some cases conflicting mechanisms31,32,33,34,35.
Caspase-8-deficient (Casp8−/−) mice or mice expressing only the DED domains of caspase-8 (Casp8DED/DED) die between embryonic day 10.5 (E10.5) and E12.5, with heart defects36,37. Culture of Casp8DED/DED embryos ex vivo rescues them from the heart defects, which suggests that the defects are a consequence, not a cause, of the underlying problem37. Indeed, mice deficient in endothelial Casp8 display the same heart abnormalities and death as those of mice with complete knockout of Casp8, which indicates that defective vasculature underlies the lethality of caspase-8 deficiency37,38. The same problems are observed in mice deficient in the Fas-associated death domain (FADD) (Fadd−/−)39. Unexpectedly, cFlip-deficient mice (Cflar−/−; called 'Cflip−/−' here), which should have increased caspase-8 activity, are ostensibly a phenocopy of Fadd−/− mice40. However, because mice can be rescued from the lethality of deficiency in caspase-8 or FADD by loss of RIPK3, it has been proposed that caspase-8 activity is needed to inhibit necroptosis at this embryonic stage and that this cell death results in the lethality observed41,42,43,44. Indeed, it may be a heterodimer of caspase-8 and cFLIPL (the long isoform of cFLIP) that retains restricted catalytic activity compared with that of the caspase-8 homodimer and can cleave RIPK1 (ref. 45) (Fig. 2) that is ultimately responsible for inhibiting RIPK3 activation42, which would neatly explain the similar in vivo phenotypes. In agreement with that proposal, the heterodimer of caspase-8 and cFLIPs (the short form of cFLIP), which is unable to create caspase-8 with restricted activity as does the caspase-8–cFLIPL heterodimer, sensitizes cells to Fas- and TLR-induced necroptosis46,47.
Analysis of the embryonic 'checkpoint' discussed above has extended to Ripk1−/− mice. Deletion of Ripk1 prevents the embryonic death of Fadd−/− and Casp8−/− mice, although Ripk1−/−Fadd−/− and Ripk1−/−Casp8−/− mice die perinatally, like Ripk1−/− mice43,48,49,50. It has also been shown that the death at E10.5 is due to TNFR1 signaling, because the loss of the gene encoding TNFR1 (Tnfr1) also inhibits the lethality of caspase-8 deficiency48. This result is fascinating. First, mice deficient in both cIAP1 and cIAP2 (Birc2−/−Birc3−/−; called 'Ciap1−/−Ciap2−/−' here) and mice deficient in HOIP, the main catalytic subunit of LUBAC (Rnf31−/−; called 'Hoip−/−' here), similarly die at E10.5, and their death at this embryonic stage is also prevented by loss of RIPK1, RIPK3 or TNFR1 (Ciap1−/−Ciap2−/−)51 or TNFR1 (Hoip−/−)20. This indicates that cIAPs are involved in this same checkpoint. Second, this checkpoint seems to show that caspase-8 is activated by TNF. The developmental purpose of such a TNF signal at E10.5 is unclear, because mice deficient in Tnf or Tnfr1 develop normally. Because cIAPs are known to inhibit RIPK1- and RIPK3-mediated necroptosis in tissue culture, these results again seem to support the idea that there is a necroptotic event in these mutant mice that causes embryonic death. It has been shown that during TNF-induced necroptosis, RIPK3 is ubiquitinated at Lys5, which supports the formation of a RIPK1-RIPK3 complex, while A20, via its deubiquitinase activity, diminishes the ubiquitination of Lys5 to inhibit necroptosis52. Interestingly, loss of RIPK3 extends the life of mice deficient in A20, which suggests that the lethality caused by A20 deficiency is partially due to excess necroptosis, a result that will require confirmation by co-deletion of MLKL.
RIPK1 in cell-death pathways
One of the most satisfying recent findings, which has confirmed considerable earlier in vitro work, is that two different mutant mice that express kinase-inactive RIPK1 (D138N and K45A) are viable, while cells from these mice are completely resistant to necroptotic stimuli50,53 (Figs. 1 and 3). Mutant cells expressing kinase-inactive RIPK1 activate NF-κB normally, produce wild-type concentrations of cytokines in response to TNF and are as resistant as wild-type cells to apoptotic stimuli50,53. Thus, the perinatal death of Ripk1−/− mice must be due to the loss of the structural role that RIPK1 serves and not loss of its kinase activity (Fig. 1). These mutant mice expressing kinase-inactive RIPK1 can now be deployed to confirm other pathological scenarios in which necrostatin (a small-molecule allosteric inhibitor of RIPK1's kinase activity) has been used.

Necroptosis typically relies on this kinase activity, as specific kinase inhibitors (Nec-1 or the GSK inhibitors of RIPK3 (RIPK3i (GSK840, GSK843, GSK872)) or kinase-inactive ('kinase-dead' (KD)) mutants block pathway activation. Genetic crosses involving RIPK1-deficient mice have revealed that RIPK1 has kinase-independent scaffold activity that is needed to inhibit cell death, as RIPK1 deficiency can activate either apoptosis or necroptosis depending on the context and cell type. Simultaneously, these crosses also show that RIPK1 is dispensable for necroptosis. Inhibition of the kinase activity of RIPK3 via genetic means (the D161N mutant of RIPK3) or chemical means can lead to the formation of a RIPK3–RIPK1–FADD–caspase-8 complex that triggers apoptosis, although this is not a universal outcome of RIPK3 kinase inhibition, as other kinase-inactive RIPK3 mutants (K51A, D161G or D143N) inhibit necroptosis without triggering apoptosis. Necroptosis can also be inhibited in human cells by treatment with the MLKL inhibitor necrosulfonamide or compound 1 (refs. 35,108) (bottom right). Ub, ubiquitin.
The fact that mutant mice expressing kinase-inactive RIPK1 survive indicates that Ripk1−/− mice die perinatally from aberrant signaling that is not due to necroptosis but is due to apoptosis, a conclusion that is consistent with the fact that both RIPK3-deficient (Ripk3−/−) mice and MLKL-deficient (Mlkl−/−) mice are viable54,55,56. Nevertheless, published work from three independent laboratories has suggested the opposite is true. Surprisingly, the perinatal lethality of RIPK1 deficiency is not prevented by loss of caspase-8 (refs. 48,49,50), although the embryonic lethality of caspase-8 deficiency is prevented by loss of RIPK1 (Fig. 1). Thus, the finding that the loss of RIPK1 acts as a phenocopy of the loss of RIPK3 in protecting Casp8−/− mice from death at E10.5 supports the idea that the E10.5 checkpoint involves a necroptotic component that requires RIPK1 for its execution. However, the fact that Ripk3−/−Casp8−/− and Ripk3−/−Fadd−/− mice survive past weaning but Ripk1−/−Casp8−/− and Ripk1−/−Fadd−/− mice do not indicates that RIPK1 has a protective role, whereas RIPK3 does not48,49,50.
RIPK1 as a necroptosis inhibitor
The fact that the loss of RIPK3 protects Ripk1−/−Casp8−/− mice raises the awkward question of whether necroptosis underlies the lethality of RIPK1 deficiency48,49,50 (Fig. 1). This is awkward because it has been widely assumed that the kinase activity of RIPK1 is required for necroptosis. Surprisingly, loss of RIPK3 or MLKL does indeed provide some protection to Ripk1−/− mice48,49,50 (Fig. 1). Both Ripk1−/−Ripk3−/− mice and Ripk1−/−Mlkl−/− mice survive longer than Ripk1−/− mice do, by a few days, but they eventually succumb to an intestinal defect marked by excessive apoptosis48,49, a finding further supported by two other studies of mice with specific deletion of RIPK1 in the intestinal epithelium57,58. Furthermore, as discussed above, loss of RIPK1's kinase activity alone cannot account for the lethality of RIPK1 deficiency, because mutant mice expressing kinase-inactive RIPK1 survive (Fig. 1). Because RIPK3 has alternative roles in inflammation and not just in effecting necroptosis, the result obtained with the Ripk1−/−Mlkl−/− cross is particularly important. Furthermore, because the pseudokinase MLKL is a terminal necroptotic effector and thus far does not seem to substantially regulate inflammatory cytokine production, we suggest that Ripk1−/− mice die perinatally due to excessive necroptosis, which is independent of the pro-necrotic kinase activity of RIPK1.
But what drives necroptosis in Ripk1−/− mice? Interferon signaling might provide a partial answer, because the survival of mice deficient in RIPK1, TNFR1 and the type I interferon receptor IFNAR (Ripk1−/−Tnfr1−/−Ifnar−/−) is extended beyond that of Ripk1−/−Tnfr1−/− mice48. Furthermore, Ripk1−/− mouse embryonic fibroblasts and Ripk1−/−Casp8−/− cells are hypersensitive to type I interferon–induced death, which is inhibited by a RIPK3 kinase inhibitor and or by knockdown of RIPK3 or MLKL48,50. Another explanation might be that necroptosis is initiated by TLR signaling via TRIF, which can activate RIPK3 via its own RHIM domain, as the survival of mice deficient in RIPK1, TNFR1 and TRIF (Ripk1−/−Tnfr1−/−Trif−/−) is similar to Ripk1−/−Tnfr1−/−Ifnar−/− mice48 (Fig. 1). That being said, because Ripk1−/−Tnfr1−/−Ifnar−/− mice and Ripk1−/−Tnfr1−/−Trif−/− mice are less viable than Ripk1−/−Ripk3−/−Casp8−/− mice are, aberrant signaling via type I interferon or the TLR is probably not the only driver of necroptosis48 (Fig. 1). It is possible that loss of signaling via type I interferon and loss of signaling via TLR together will afford full protection, but the quadruple-knockout cross may be 'an experimental bridge too far'. A further intriguing possibility is that RIPK1 directly inhibits autonomous activation of RIPK3. In support of this idea, it has been demonstrated that RIPK1 inhibits autoactivation of a version of RIPK3 that can artificially form dimers59. Consistent with the data obtained with knockout mice and the idea that RIPK1 inhibits the activation of RIPK3, it has also been demonstrated that cells with knockdown of RIPK1 are actually more sensitive to necroptosis than are wild-type cells in response to both TNF-induced necroptosis and TLR-induced necroptosis60. On the basis of these results, it has been proposed that necrostatin, rather than preventing the activation of necroptosis by RIPK1, might block necroptosis by enhancing RIPK1's inhibitory properties60.
Multiple roles for RIPK1 and RIPK3
The fact that loss of the key necroptotic-cell-death effectors RIPK1 and RIPK3 prevents the early lethality of caspase-8 deficiency is not as helpful in resolving the Casp8−/−–versus–Cflip−/− enigma as it first appears, because RIPK1 and RIPK3 have roles in TLR signaling pathways, inflammasome signaling pathways and other inflammatory signaling pathways: RIPK1 is a pivotal scaffold element in several inflammatory pathways, and RIPK3 probably has many other substrates in addition to MLKL (Fig. 4). For example, treatment of bone marrow–derived macrophages (BMDMs) with a peptido-mimetic of the natural IAP ('inhibitor of apoptosis') antagonist Smac (DIABLO) results in depletion and antagonism of IAPs and induces the secretion of TNF and other inflammatory cytokines, and this is largely prevented by inhibition of RIPK1 or deletion of Ripk3 (ref. 61). Similarly, loss of caspase-8 primes the MAVS–RIG-I complex to respond to viral infection in a RIPK1-dependent manner62. Furthermore, treatment of BMDMs or bone marrow–derived dendritic cells (BMDCs) with a Smac mimetic results in activation of the NLRP3 inflammasome or activation of caspase-8 and secretion of IL-1β that is RIPK3 dependent but MLKL independent63,64. Similar to cells deficient in the apoptosis inhibitors XIAP, cIAP1 and cIAP2 (ref. 63), Casp8−/− BMDCs have a constitutively active form of the NLRP3 inflammasome whose activity depends on RIPK3 (ref. 65) (Fig. 4). These results suggest that IAPs and caspase-8 inhibit RIPK3-dependent activation of the NLRP3 inflammasome. In contradiction of that interpretation, other studies have shown that loss of caspase-8 results in diminished inflammasome function, because Fadd−/−Ripk3−/− and Casp8−/−Ripk3−/− BMDMs are defective in their ability to activate the inflammasome, but Ripk3−/− BMDMs are not66. Complicating the situation further still, caspase-8 can bypass NLRP inflammasomes and caspase-1 and directly process IL-1β into its active inflammatory form following treatment with stimuli such as Smac mimetics63, chemotherapeutic drugs67 and FasL68 (Fig. 4). Thus, loss of caspase-8 can potentially simultaneously lead to an increase in inflammasome activation and reduction in IL-1β-processing power. Inflammasomes in Ripk1−/− BMDMs, like those in Casp8−/− BMDCs65, have been shown to be in a mildly activated state, and this is dependent upon the presence of RIPK3 (ref. 49); however, there is little evidence of inflammasome activation or activity in Ripk1−/− mice49. The strongest argument that the early embryonic death of Casp8−/− mice is indeed due to necroptosis is the fact that it has been shown that MLKL deficiency prevents this death (Fig. 1; S. Alvarez-Diaz and A. Strasser, personal communication).

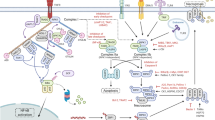
Marina Corral Spence/Nature Publishing Group
Necroptotic DAMPs can initiate a secondary inflammatory response in neighboring cells in a manner dependent on MyD88 and NF-κB: HMGB1, via TLR4 and RAGE; IL-1α, via IL-1R; and IL-33, via IL-1R and ST2. HMGB1-TLR4 can be internalized into the endosome to initiate, like the viral double-stranded RNA–TLR3 pathway, two different TRIF-dependent pathways. The first, a RIPK1-dependent pathway, leads to NF-κB-induced pro-inflammatory cytokine production (bottom left). The second pathway involves induction of IRF7 and production of IFN-b. Mitochondrial DNA leads to Myd88-dependent TLR9 signaling with induction of NF-κB or IRF. TNF triggers NF-κB signaling that is subject to regulation by RIPK1 (Fig. 2). Loss of cIAPs can lead to TNF-induced phosphorylation of RIPK3 or activation of caspase-8. Phosphorylation of RIPK3 can lead to formation of the NLRP3-inflammasome (middle right) or pro-inflammatory cytokine production via an unknown mechanism (dashed black arrow). The NLRP3 inflammasome activates caspase-1 that generates mature IL-1β. Furthermore, this inflammasome can be directly activated by the necroptotic DAMP ATP via the receptor P2RX7. Active caspase-8 can either cleave IL-1β to its mature form directly or do so by activating NLRP3-inflammasome via an unknown mechanism (dashed arrows). Furthermore, caspase-8 can cleave RIPK1 to inhibit activation of the transcription factor IRF3 by RIG-I (middle). The release of pro-inflammatory cytokines and interferons can lead to the killing (Fig. 2) or recruitment of infiltrating cells of the immune system or to further death of neighboring cells, which promotes further inflammation.
Clearly this is an emerging area, with some seemingly contradictory results, but the simplest 'take-home' message from these studies is that loss of upstream components of the cell-death signaling axis, including RIPK1, RIPK3 and caspase-8, probably affects other inflammatory signaling pathways in ways that are not yet fully understood, and their loss may induce or reduce inflammatory signaling. However, in the absence of strong genetic evidence to the contrary, it is unlikely that downstream apoptosis or necroptosis effectors such as CAD ('caspase-activated DNase') or MLKL serve multiple roles.
Evidence for necroptotic DAMPs
Why do Ripk1−/− mice die around birth? The most striking feature of Ripk1−/− mice is systemic and multi-organ inflammation, which is diminished in Ripk1−/−Ripk3−/− mice and Ripk1−/−Mlkl−/− mice49 (Figs. 1 and 5). Furthermore deletion of the gene encoding MyD88 (Myd88), a key adaptor in DAMPs' signaling through TLRs, as well as the receptor RAGE, the receptor IL-1R1 and heterodimeric receptor Il-1R–ST2L69 also diminishes the inflammation of Ripk1−/− mice49 (Fig. 1). These results suggest that necroptosis-induced DAMPs drive the inflammation and death of Ripk1−/− mice. In agreement with that, skin inflammation in Ripk1−/− mice and mice with epidermis-specific RIPK1 deficiency is prevented by loss of MLKL or RIPK3 (necroptosis) but not by deletion of FADD (apoptosis)48,49,57. There are several proposed DAMPs that signal through TLRs, such as HMGB1, mitochondrial DNA and HSPs; however, HMGB1 levels are not higher in the plasma of Ripk1−/− mice than in that of wild-type mice49. Myd88 deficiency can also inhibit IL-1β signaling, but there is likewise little evidence for processing of IL-1β by caspase-1 in Ripk1−/− mice49. However, IL-1α and IL-33, which also signal through the family of IL-1 receptors, are readily detected in the plasma of Ripk1−/− mice but not in that of Ripk1−/−Ripk3−/− or Ripk1−/−Mlkl−/− mice, which suggests that necroptosis-induced release of IL-1α and IL-33 may drive inflammatory disease in vivo49. IL-33 is a particularly interesting molecule, because it is a chromatin-associated protein in healthy cells, is mostly constitutively expressed in epithelial and endothelial cells70 and does not seem to be conventionally secreted but is instead released from dying cells. Furthermore, it is inactivated by the apoptotic effectors caspase-3 and caspase-7 (refs. 71,72), which fulfills many of the requirements of an ideal DAMP: activated following a dangerous death, but inactivated by a scheduled apoptotic death. Interestingly, IL-33 can also be processed by proteases present in neutrophils, and cleavage by these proteases increases the activity of IL-33 approximately tenfold73. In correlation with their large number of neutrophils and high levels of inflammation, Ripk1−/− and Ripk1−/−Casp8−/− mice have this active cleaved form of IL-33 in their plasma, but Ripk1−/−Ripk3−/− and Ripk1−/−Mlkl−/− mice do not49.

Marina Corral Spence/Nature Publishing Group
A plethora of evidence, mainly from mouse models of disease, indicates that RIPK1, RIPK3 and MLKL are important mediators of tissue damage. Delineating the relative contributions of these regulators to cell death versus inflammatory cytokine production in these disease processes has proven extremely challenging. While findings from these models have been encouraging, whether they translate to human disease largely remains to be seen.
RIP kinases and MLKL as therapeutic targets
In addition to being able to promote developmental and sterile inflammation, RIPK1 has been linked to a pathogenic role in multiple tissue-damage models, including heart attack, atherosclerosis, pancreatitis, and liver, retinal and renal injury (Fig. 5). However, whether RIPK1 mediates damage via the induction of necroptosis in all these models is unclear, particularly if such a conclusion is based only on an effect of necrostatin-1 (Nec-1). This is highlighted by work showing that crossing of Sharpin mutant mice (with the spontaneous mutation cpdm) to Ripk1K45A/K45A mice (which express kinase-inactive RIPK1) completely prevents the multi-organ inflammatory phenotype that results from the cpdm mutation74. While many features of the systemic phenotype that results from the cpdm mutation have been shown to be dependent on both RIPK3 and MLKL, the hallmark dermatitis of these mice is not75,76. Instead, the dermatitis is caused mainly by apoptosis dependent on TNFR1, TRADD and caspase-8, which suggests that in this case, the kinase activity of RIPK1 'sits' upstream of caspase-8 (refs. 75,76). This is a fascinating finding and is reminiscent of the various tissue-specific roles of RIPK1 demonstrated in Ripk1−/− mice49. Furthermore, the result highlights the pitfalls of claiming that a phenotype is driven by necroptosis. Thus, in the absence of confirmatory data obtained with mice deficient in MLKL (and in RIPK3, where it overlaps), it is prudent to be circumspect about such conclusions. Some confirmation of the Nec-1 result is provided by the finding that Ripk1D138N/D138N mice (which express kinase-inactive RIPK1) and Ripk3−/− mice are similarly protected from TNF-induced hypothermia53,77 (Fig. 5). Similarly, Nec-1 increases survival after injury in an ischemia-reperfusion model that simulates renal damage following kidney transplantation78, findings recapitulated in Ripk3−/− mice79. However, again, the effect of loss of RIPK3 on reducing kidney ischemia-reperfusion injury may not be due to loss of necroptosis, as first assumed, but may be due to a greater kidney peritubular diameter in Ripk3−/− mice than in wild-type mice80. The fact that RIPK1 appears able to induce caspase-8-dependent apoptosis in some circumstances might mean Nec-1 is even more potent than previously thought, by virtue of this newfound versatility. However, Nec-1 is a far-from-ideal drug, having a very short in vivo half-life and nonspecific activity against IDO (indoleamine 2,3-dioxygenase), an enzyme also involved in the inflammatory response81. The more-specific RIPK1 inhibitor Nec-1s is a better proposition but probably has similarly poor pharmacokinetic properties. Regardless of these caveats concerning existing drugs directed against RIPK1, the fact that Ripk1D138N/D138N mice and Ripk1K45A/K45A mice have no overtly altered phenotype50,53,74 suggests that the kinase activity of RIPK1 may be a safe therapeutic target.
Whether targeting the kinase activity of RIPK3 will have a utility similar to targeting RIPK1 is less clear. The fact that within 2 days of the induction of a Ripk3D161N/D161N knock-in mutation that results in kinase-inactive RIPK3, adult mice die of rapid weight loss and excessive intestinal cell death raises concern53, although a subsequent study has shown that a different mutant mouse expressing kinase-inactive RIPK3, the Ripk3K51A/K51A mouse, is viable82. Furthermore, this subsequent study82 has shown that the D161N mutant form of RIPK3 induces apoptosis and that treatment with various inhibitors of RIPK3 is also able achieve the same result in wild-type cells (Fig. 3), probably because of a conformational change induced in RIPK3 that allows it to activate caspase-8 via RIPK1. Similarly, a study has suggested that wild-type RIPK3 may undergo a similar conformation change to induce TRIF-dependent activation of caspase-8 and processing and secretion of IL-1β83. This effect clearly limits the potential utility of RIPK3 kinase inhibitors in blocking cell death. One insight obtained from studies described above of mice with a combination of knockout and knock-in mutations, and particularly mice with triple-knockout mutations, is that targeting necroptosis alone will probably not be a universal therapeutic panacea and a better strategy might involve concurrent inhibition of caspase-8.
RIP kinases during infection
Given RIPK1 has an important structural role in inflammatory cytokine production induced by TNF, TLRs and NF-κB (Fig. 4), it is unsurprising that RIPK1 is functionally linked to many infection scenarios84. In addition to being important in those pathways, RIPK1 is also important for the expression of type I interferon in response to single-stranded RNA viruses62,85 (Fig. 4). In uninfected cells, caspase-8 seems to limit pathway activation by cleaving RIPK1, because caspase-8-deficient cells are 'primed' for IRF3 activation62, a finding that suggests a mechanism for the chronic inflammation observed in mice with conditional deletion of caspase-8 (ref. 62) (Fig. 4).
As discussed above, host cells activate apoptosis to limit viral replication, often via pathways dependent on RIPK1 and caspase-8. An early indication that programmed necrosis might be important in antiviral defense was provided by work with vaccinia virus, a double-stranded DNA virus that can inhibit apoptosis86,87 and pyroptosis88. RIPK1 has been shown to be required for TNF-induced necrosis of vaccinia virus–infected cells89, a process subsequently found to require RIPK3 (ref. 90). The strongest argument for the importance of necroptosis during viral infection has been provided by studies of murine cytomegalovirus, another double-stranded DNA virus. Murine cytomegalovirus encodes a protein, vIRA (M45), that interacts via its RHIM domain with both RIPK1 and RIPK3 to inhibit DAI-dependent necroptosis91,92,93. Furthermore, it appears that human cytomegalovirus adopts a different strategy to inhibit necroptosis downstream of the phosphorylation of RIPK3 and MLKL via a yet-to-be-discovered mechanism94. Necroptosis may also occur in CD4+ T cells infected with human immunodeficiency virus, because treatment with Nec-1 or with the human MLKL inhibitor necrosulfonamide (Fig. 3) restrains this cytopathic effect of human immunodeficiency virus95. A direct interaction of the RHIM domains of RIPK1 and RIPK3 with the herpes simplex virus type 1 protein ICP6 and herpes simplex virus type 2 protein ICP10 has been shown to induce RIPK3-MLKL–dependent necroptosis in mouse cells that consequently leads to a host antiviral response96,97. Interestingly, the same herpes simplex virus proteins have been shown to inhibit TNF-induced necroptosis in human cells, which indicates the importance of specific host factors97,98.
Bacterial infection can also induce cell death dependent on RIPK1 and RIPK3. Salmonella enterica serovar Typhimurium can induce macrophage pyroptosis and has been found to induce a necroptotic autocrine loop via production of type I interferon that subsequently triggers RIPK3-dependent necroptosis99 (Fig. 3). Bacteria have also evolved to inhibit RIPK1 signaling; for example, Porphyromonas gingivalis has been shown to specifically cleave RIPK1 via its lysine-specific (Kgp) protease100.
Although RIP kinases are clearly important during infection, disentangling relative contributions from direct transcriptional versus cell-death functions is still one of the most challenging issues in this field, analogous to caspase-1's role in cytokine production versus its role in pyroptotic cell death101. For example, on the one hand, Yersinia pestis can inhibit NF-κB, which leads to degradation of cFLIP, activation of caspase-8, apoptosis, and cleavage of RIPK1 via a platform dubbed the 'ripoptosome'47,102. On the other hand, a RIPK1–caspase-8 pathway is required for Y. pestis infection–induced production of TNF and IL-6 and processing of IL-1β and IL-18 by caspase-1 (refs. 103,104). This leaves open the question of what is more physiologically important: the contribution of RIPK1 to transcription or its contribution to cell death.
Promising, but unproven
Fundamental questions in the field of necroptosis research are how, where and when it occurs. The lack of markers of necroptosis has hampered the ability to address the 'where' and 'when' questions, although an antibody to phosphorylated MLKL might go some way toward addressing this void, at least in humans32. Such tools should enable better investigation of disease processes that involve necroptosis in vivo and should definitively address whether necroptosis is a worthwhile therapeutic target. Certainly, results obtained with the extensive genetic crosses described in this Review (Fig. 1) provide strong support for the hypothesis that there is a necroptotic component to certain inflammatory diseases. The 'how' question turns out to have some intriguing answers, and studies discussed in this Review have almost completely laid to rest the longstanding mystery of why Ripk1−/− mice die and have simultaneously identified unexpected roles for RIPK1 in inhibiting necroptosis while confirming other known roles. They suggest that RIPK3 may also have roles in inhibiting apoptosis and have provided some, albeit contradictory, data to suggest how MLKL kills necroptotic cells. They certainly provide more than enough justification for going back over old cases2 to look for new insights48,49,50.
Change history
26 June 2015
In the version of this article initially published, the following were incorrect and should be corrected as stated here. In Fig 1, ref. 84 should be ref. 82, and "pers. comm." should be cited one line below (aligned with Mlkl-/-). The middle of the third sentence of the Fig. 2 legend should include not a period but a comma, to read "phosphorylation of RIPK3, which leads to" and the end of fifth sentence should not include "RIPK" but should read "which leads to RIPK1-independent necroptosis...." The fifth sentence of final paragraph of the subsection "RIPK1-, RIPK3- and MLKL-induced necroptosis" should not include "cIAP" but should begin "First, mice deficient in both cIAP1 and cIAP2...." The middle of the sixth sentence of first paragraph in the subsection "Evidence for necroptotic DAMPS" should not state this as "concentration" but should read "however, HMGB1 levels are not higher" and final sentence should include citation of ref. 49 to read "...do not49." In Fig. 4, the arrow from pro-IL-1β should be a solid line, not a dashed line. The third sentence of the first paragraph of the subsection "RIP kinases and MLKL as therapeutic targets" should cite ref. 74 instead of ref. 76 (to read "mutation74."), and the final sentence of that subsection should be deleted. Fig. 5 should include the following revisions: in the top right section (Brain), the text should not include a parenthetical element but read "brain injury117,118, controlled cortical impact trauma119 and"; in the upper left section (Skin), the first sentence should not end with "RIPK" but should end "RIPK1 (refs. 48,49,57)"; in the middle left section (Liver), "no effect on" should be orange in both places (not only the first); and in the bottom section (Systemic), the second line should read "TNFand Z-VAD-induced hyper-acute shock" (not "TNF- or Z-VAD-"). The errors have been corrected in the HTML and PDF versions of the article.
References
Galluzzi, L. et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 22, 58–73 (2015).
Kelliher, M.A. et al. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8, 297–303 (1998).
Wallach, D., Kang, T.B. & Kovalenko, A. Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat. Rev. Immunol. 14, 51–59 (2014).
Matzinger, P. An innate sense of danger. Semin. Immunol. 10, 399–415 (1998).
Cullen, S.P. et al. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol. Cell 49, 1034–1048 (2013).
Vaux, D.L., Haecker, G. & Strasser, A. An evolutionary perspective on apoptosis. Cell 76, 777–779 (1994).
Gallucci, S., Lolkema, M. & Matzinger, P. Natural adjuvants: endogenous activators of dendritic cells. Nat. Med. 5, 1249–1255 (1999).
Taylor, R.C., Cullen, S.P. & Martin, S.J. Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 9, 231–241 (2008).
Mukae, N., Yokoyama, H., Yokokura, T., Sakoyama, Y. & Nagata, S. Activation of the innate immunity in Drosophila by endogenous chromosomal DNA that escaped apoptotic degradation. Genes Dev. 16, 2662–2671 (2002).
Kawane, K., Tanaka, H., Kitahara, Y., Shimaoka, S. & Nagata, S. Cytokine-dependent but acquired immunity-independent arthritis caused by DNA escaped from degradation. Proc. Natl. Acad. Sci. USA 107, 19432–19437 (2010).
Feldmann, M. & Maini, R.N. Anti-TNF therapy, from rationale to standard of care: what lessons has it taught us? J. Immunol. 185, 791–794 (2010).
Dinarello, C.A., Simon, A. & van der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 11, 633–652 (2012).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Croker, B.A., O'Donnell, J.A. & Gerlic, M. Pyroptotic death storms and cytopenia. Curr. Opin. Immunol. 26, 128–137 (2014).
Chen, K.W. et al. The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 8, 570–582 (2014).
Gross, O. et al. Inflammasome activators induce interleukin-1a secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36, 388–400 (2012).
Silke, J. The regulation of TNF signalling: what a tangled web we weave. Curr. Opin. Immunol. 23, 620–626 (2011).
Wertz, I.E. & Dixit, V.M. Regulation of death receptor signaling by the ubiquitin system. Cell Death Differ. 17, 14–24 (2010).
Silke, J. & Brink, R. Regulation of TNFRSF and innate immune signalling complexes by TRAFs and cIAPs. Cell Death Differ. 17, 35–45 (2010).
Peltzer, N. et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 9, 153–165 (2014).
Silke, J. & Vaux, D.L. IAP gene deletion and conditional knockout models. Semin. Cell Dev. Biol. 39, 97–105 (2015).
Benedict, C.A., Banks, T.A. & Ware, C.F. Death and survival: viral regulation of TNF signaling pathways. Curr. Opin. Immunol. 15, 59–65 (2003).
Silke, J. & Hartland, E.L. Masters, marionettes and modulators: intersection of pathogen virulence factors and mammalian death receptor signaling. Curr. Opin. Immunol. 25, 436–440 (2013).
Bonnet, M.C. et al. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 35, 572–582 (2011).
Gerlach, B. et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591–596 (2011).
Hitomi, J. et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311–1323 (2008).
Wang, L., Du, F. & Wang, X. TNF-a induces two distinct caspase-8 activation pathways. Cell 133, 693–703 (2008).
Li, J. et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 (2012).
Mocarski, E.S., Upton, J.W. & Kaiser, W.J. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat. Rev. Immunol. 12, 79–88 (2012).
Moujalled, D.M., Cook, W.D., Murphy, J.M. & Vaux, D.L. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis. 5, e1086 (2014).
Chen, X. et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 24, 105–121 (2014).
Wang, H. et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146 (2014).
Cai, Z. et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 16, 55–65 (2014).
Dondelinger, Y. et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 7, 971–981 (2014).
Hildebrand, J.M. et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. USA 111, 15072–15077 (2014).
Varfolomeev, E.E. et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9, 267–276 (1998).
Sakamaki, K. et al. Ex vivo whole-embryo culture of caspase-8-deficient embryos normalize their aberrant phenotypes in the developing neural tube and heart. Cell Death Differ. 9, 1196–1206 (2002).
Kang, T.B. et al. Caspase-8 serves both apoptotic and nonapoptotic roles. J. Immunol. 173, 2976–2984 (2004).
Yeh, W.C. et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279, 1954–1958 (1998).
Yeh, W.C. et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 12, 633–642 (2000).
Kaiser, W.J. et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 (2011).
Oberst, A. et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 (2011).
Zhang, H. et al. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 471, 373–376 (2011).
Dillon, C.P. et al. Survival function of the FADD-CASPASE-8-cFLIP(L) complex. Cell Rep. 1, 401–407 (2012).
Pop, C. et al. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem. J. 433, 447–457 (2011).
Geserick, P. et al. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 187, 1037–1054 (2009).
Feoktistova, M. et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 (2011).
Dillon, C.P. et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202 (2014).
Rickard, J.A. et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157, 1175–1188 (2014).
Kaiser, W.J. et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci. USA 111, 7753–7758 (2014).
Moulin, M. et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 31, 1679–1691 (2012).
Onizawa, M. et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 16, 618–627 (2015).
Newton, K. et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360 (2014).
Newton, K., Sun, X. & Dixit, V.M. Kinase RIP3 is dispensable for normal NF-κBs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell. Biol. 24, 1464–1469 (2004).
Wu, J. et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 23, 994–1006 (2013).
Murphy, J.M. et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453 (2013).
Dannappel, M. et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94 (2014).
Takahashi, N. et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature 513, 95–99 (2014).
Orozco, S. et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 21, 1511–1521 (2014).
Kearney, C.J., Cullen, S.P., Clancy, D. & Martin, S.J. RIPK1 can function as an inhibitor rather than an initiator of RIPK3-dependent necroptosis. FEBS J. 281, 4921–4934 (2014).
Wong, W.W. et al. cIAPs and XIAP regulate myelopoiesis through cytokine production in a RIPK1 and RIPK3 dependent manner. Blood 123, 2562–2572 (2014).
Rajput, A. et al. RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity 34, 340–351 (2011).
Vince, J.E. et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36, 215–227 (2012).
Lawlor, K.E. et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6, 6282 (2015).
Kang, T.B., Yang, S.H., Toth, B., Kovalenko, A. & Wallach, D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38, 27–40 (2013).
Gurung, P. et al. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 192, 1835–1846 (2014).
Antonopoulos, C., El Sanadi, C., Kaiser, W.J., Mocarski, E.S. & Dubyak, G.R. Proapoptotic chemotherapeutic drugs induce noncanonical processing and release of IL-1β via caspase-8 in dendritic cells. J. Immunol. 191, 4789–4803 (2013).
Bossaller, L. et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 189, 5508–5512 (2012).
Kaczmarek, A., Vandenabeele, P. & Krysko, D.V. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223 (2013).
Palmer, G. & Gabay, C. Interleukin-33 biology with potential insights into human diseases. Nat. Rev. Rheumatol. 7, 321–329 (2011).
Lüthi, A.U. et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31, 84–98 (2009).
Cayrol, C. & Girard, J.P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. USA 106, 9021–9026 (2009).
Lefrançais, E. et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA 109, 1673–1678 (2012).
Berger, S.B. et al. Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol. 192, 5476–5480 (2014).
Kumari, S. et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. eLife 3, e03422 (2014).
Rickard, J.A. et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife 3, e03464 (2014).
Duprez, L. et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35, 908–918 (2011).
Linkermann, A. et al. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 81, 751–761 (2012).
Lau, A. et al. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am. J. Transplant. 13, 2805–2818 (2013).
Linkermann, A. et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl. Acad. Sci. USA 111, 16836–16841 (2014).
Vandenabeele, P., Grootjans, S., Callewaert, N. & Takahashi, N. Necrostatin-1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ. 20, 185–187 (2013).
Mandal, P. et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 56, 481–495 (2014).
Moriwaki, K., Bertin, J., Gough, P.J. & Chan, F.K.A. RIPK3-caspase 8 complex mediates atypical pro-IL-1β processing. J. Immunol. 194, 1938–1944 (2015).
Christofferson, D.E., Li, Y. & Yuan, J. Control of life-or-death decisions by RIP1 kinase. Annu. Rev. Physiol. 76, 129–150 (2014).
Balachandran, S., Thomas, E. & Barber, G.N. A FADD-dependent innate immune mechanism in mammalian cells. Nature 432, 401–405 (2004).
Dobbelstein, M. & Shenk, T. Protection against apoptosis by the vaccinia virus SPI-2 (B13R) gene product. J. Virol. 70, 6479–6485 (1996).
Wasilenko, S.T., Stewart, T.L., Meyers, A.F. & Barry, M. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc. Natl. Acad. Sci. USA 100, 14345–14350 (2003).
Gerlic, M. et al. Vaccinia virus F1L protein promotes virulence by inhibiting inflammasome activation. Proc. Natl. Acad. Sci. USA 110, 7808–7813 (2013).
Chan, F.K. et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J. Biol. Chem. 278, 51613–51621 (2003).
Cho, Y.S. et al. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009).
Upton, J.W., Kaiser, W.J. & Mocarski, E.S. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J. Biol. Chem. 283, 16966–16970 (2008).
Upton, J.W., Kaiser, W.J. & Mocarski, E.S. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7, 302–313 (2010).
Upton, J.W., Kaiser, W.J. & Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 (2012).
Omoto, S. et al. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J. Biol. Chem. 290, 11635–11648 (2015).
Pan, T. et al. Necroptosis takes place in human immunodeficiency virus type-1 (HIV-1)-infected CD4+ T lymphocytes. PLoS ONE 9, e93944 (2014).
Wang, X. et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. USA 111, 15438–15443 (2014).
Huang, Z. et al. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 17, 229–242 (2015).
Guo, H. et al. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 17, 243–251 (2015).
Robinson, N. et al. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 13, 954–962 (2012).
Madrigal, A.G., Barth, K., Papadopoulos, G. & Genco, C.A. Pathogen-mediated proteolysis of the cell death regulator RIPK1 and the host defense modulator RIPK2 in human aortic endothelial cells. PLoS Pathog. 8, e1002723 (2012).
Croker, B.A., O'Donnell, J.A. & Gerlic, M. Pyroptotic death storms and cytopenia. Curr. Opin. Immunol. 26, 128–137 (2014).
Grobner, S. et al. Catalytically active Yersinia outer protein P induces cleavage of RIP and caspase-8 at the level of the DISC independently of death receptors in dendritic cells. Apoptosis 12, 1813–1825 (2007).
Weng, D. et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl. Acad. Sci. USA 111, 7391–7396 (2014).
Philip, N.H. et al. Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling. Proc. Natl. Acad. Sci. USA 111, 7385–7390 (2014).
Cusson., N, Oikemus, S., Kilpatrick, E.D., Cunningham, L. & Kelliher, M. The death domain kinase RIP protects thymocytes from tumor necrosis factor receptor type 2-induced cell death. J. Exp. Med. 196, 15–26 (2002).
Roderick, J.E. et al. Hematopoietic RIPK1 deficiency results in bone marrow failure caused by apoptosis and RIPK3-mediated necroptosis. Proc. Natl. Acad. Sci. USA 111, 14436–14441 (2014).
Dowling, J.P., Nair, A. & Zhang, J. A novel function of RIP1 in postnatal development and immune homeostasis by protecting against RIP3-dependent necroptosis and FADD-mediated apoptosis. Front. Cell. Dev. Biol. 3, 12 (2015).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Murakami, Y. et al. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc. Natl. Acad. Sci. USA 109, 14598–14603 (2012).
Murakami, Y. et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 21, 270–277 (2014).
Sato, K. et al. Receptor interacting protein kinase-mediated necrosis contributes to cone and rod photoreceptor degeneration in the retina lacking interphotoreceptor retinoid-binding protein. J. Neurosci. 33, 17458–17468 (2013).
Weinlich, R. et al. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep. 5, 340–348 (2013).
Roychowdhury, S., McMullen, M.R., Pisano, S.G., Liu, X. & Nagy, L.E. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 57, 1773–1783 (2013).
Vucur, M. et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell Rep. 4, 776–790 (2013).
Welz, P.S. et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477, 330–334 (2011).
Lin, J. et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 3, 200–210 (2013).
Degterev, A. et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 (2005).
Northington, F.J., Chavez-Valdez, R., Graham, E.M., Razdan, S., Gauda, E.B. & Martin, L.J. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J. Cereb. Blood Flow Metab. 31, 178–189 (2011).
You, Z. et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 28, 1564–1573 (2008).
Zhu, S., Zhang, Y., Bai, G. & Li, H. Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington's disease. Cell Death Dis. 2, e115 (2011).
Oerlemans, M.I. et al. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res. Cardiol. 107, 270 (2012).
Smith, C.C., Davidson, S.M., Lim, S.Y., Simpkin, J.C., Hothersall, J.S. & Yellon, D.M. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc. Drugs Ther. 21, 227–233 (2007).
Linkermann, A. et al. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-alpha-induced shock. Mol. Med. 18, 577–586 (2012).
He, S. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111 (2009).
Linkermann, A. et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 110, 12024–12029 (2013).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
J.S. is involved with Catalyst Therapeutics, which is trying to develop a drug targeting MLKL for use in inflammatory diseases.
Rights and permissions
About this article

Cite this article
Silke, J., Rickard, J. & Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol 16, 689–697 (2015). https://doi.org/10.1038/ni.3206
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.3206
This article is cited by
-
The Complexity of Being A20: From Biological Functions to Genetic Associations
Journal of Clinical Immunology (2024)
-
A necroptosis-independent function of RIPK3 promotes immune dysfunction and prevents control of chronic LCMV infection
Cell Death & Disease (2023)
-
SARS-CoV-2 Z-RNA activates the ZBP1-RIPK3 pathway to promote virus-induced inflammatory responses
Cell Research (2023)
-
OASL phase condensation induces amyloid-like fibrillation of RIPK3 to promote virus-induced necroptosis
Nature Cell Biology (2023)
-
Potential therapeutic value of necroptosis inhibitor for the treatment of COVID-19
European Journal of Medical Research (2022)
