
Abstract
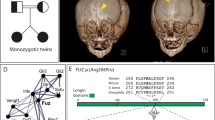
Pfeiffer syndrome (PS; McKusick MIM 101600) is an autosomal dominant craniosynostosis syndrome with characteristic craniofacial anomalies and broad thumbs and big toes1,2. We have previously demonstrated genetic heterogeneity in PS and mapped a gene to chromosome 8 (ref. 3) and a second to chromosome 10 (ref. 4). The gene on chromosome 8 is the fibroblast growth factor receptor 1 (FGFR1) with a common mutation (C755G) predicting a Pro252Arg substitution5. The gene on chromosome 10 is FGFR2 with several different mutations causing sporadic and familial PS 4,6,7,8 (Table 1). We report a recurrent single point mutation in the FGFR3 gene, located on chromosome 4p, in ten unrelated families with craniosynostosis syndromes. This mutation (C749G) predicts a Pro250Arg amino acid substitution in the extracellular domain of the FGFR3 protein. Interestingly, this common mutation occurs precisely at the analogous position within the FGFR3 protein as the mutations in FGFR1 (Pro252Arg) and FGFR2 (Pro253Arg) previously reported in Pfeiffer5 and Apert9 syndromes, respectively.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others


References
McKusick, V.A. (1994) Mendelian Inheritance in Man. A Catalog of Human Genes and Genetic Disorders. 11th edn. 759–761 (Johns Hopkins University Press, Baltimore, 1994).
Cohen, M.M. Jr., Pfeiffer syndrome update, clinical subtypes and guidelines for differential diagnosis. Am J. Med. Genet. 45, 300–307 (1993).
Robin, N.H. et al. Linkage of Pfeiffer syndrome to chromosome 8 centromere and evidence for genetic heterogeneity. Hum Mol. Genet. 3, 2153–2158 (1994).
Schell, U. et al. Mutations in FGFR1 and FGFR2 cause familial and sporadic Pfeiffer syndrome. Hum. Mol. Genet. 4, 323–328 (1995).
Muenke, M. et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nature Genet. 8, 269–274 (1994).
Lajeunie, E. et al. FGFR2 mutations in Pfeiffer syndrome. Nature Genet. 9, 108 (1995).
Rutland, P. et al. Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes. Nature Genet. 9, 173–176 (1995).
Meyers, G.A. et al. FGFR2 Exon IIIa and IIIc mutations in Crouzon, Jackson-Weiss and Pfeiffer syndromes: evidence for missense changes, insertions and a deletion due to alternative RNA splicing. Am. J. Hum. Genet. 58, 491–498 (1996).
Wilkie, A.O.M. et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nature Genet. 9, 165–172 (1995).
Hollway, G.E., Phillips, H.A., Ades, L.C., Haan, E.A. & Mulley, J.C. Localization of craniosynostosis Adelaide type to 4p16. Hum. Mol. Genet. 4, 681–683 (1995).
Von Gernet, S. et al. Craniosynostosis suggestive of Saethre-Chotzen syndrome: clinical description of a large kindred and exclusion of candidate regions on 7p. Am. J. Med. Genet. 63, 177–184 (1996).
Shiang, R. et al. Mutations in the transmembrane domain of FGFR-3 cause the most common genetic form of dwarfism, achondroplasia. Cell 78, 335–342 (1994).
Rousseau, F. et al. Mutations in the gene encoding fibrobiast growth factor receptor-3 in achondroplasia. Nature 371, 252–254 (1994).
Tavormina, P.L. et al. Thanatophoric dysplasia (types I & II) caused by distinct mutations in fibroblast growth factor receptor 3. Nature Genet. 9, 321–328 (1995).
Bellus, G.A. et al. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nature Genet. 10, 357–59 (1995).
Meyers, G.A. et al. Fibroblastgrowthfactorreceptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nature Genet. 11, 462–464 (1995).
Neilson, K.M. & Friesel, R.E. Constitutive activation of fibroblast growth factor receptor-2 by a point mutation associated with Crouzon syndrome. J. Biol. Chem. 44, 26037–26040 (1995).
Webster, M.K. & Donohue, D.J. Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO J. 15, 520–527 (1996).
Naski, M.C., Wang, Q., Xu, J. & Ornitz, D.M. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nature Genet. 13: 233–237 (1996).
Colvin, J.S., Bonne, B.A., Harding, G.W., McEwen, D.G. & Ornitz, D.M. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptors. Nature Genet. 12, 390–397 (1996).
Deng, C., Wynshaw-Boris, A., Zhou, F., Kuo, A. & Leder, P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 84, 911–921 (1996).
Johnson, D. & Williams, L.T. Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res. 60, 1–41 (1993).
Mason, I.J. The ins and outs of fibroblast growth factor receptors. Cell 78, 547–552 (1994).
Peters, K., Ornitz, D., Werner, S. & Williams, L.T. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Devl. Biol. 155, 423–430 (1993).
Li, X. et al. Effect on splicing of a silent FGFR2 mutation in Crouzon syndrome. Nature Genet. 9, 232 (1995).
Tavormina, P.L. et al. Another mutation that results in the substitution of an unpaired cysteine residue in the extracellular domain of FGFR3 in thanatophoric dysplasia. Hum. Mol. Genet. 11, 2175–2177 (1995).
Rousseau, F. et al. Missense FGFR3 mutations create cysteine residues in thanatophoric dwarfism type I (TDI). Hum. Mol. Genet 5, 509–512 (1996).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Bellus, G., Gaudenz, K., Zackai, E. et al. Identical mutations in three different fibroblast growth factor receptor genes in autosomal dominant craniosynostosis syndromes. Nat Genet 14, 174–176 (1996). https://doi.org/10.1038/ng1096-174
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/ng1096-174
