
Abstract
Pseudoautosomal region 1 (PAR1) contains SHOX, in addition to seven highly conserved non-coding DNA elements (CNEs) with cis-regulatory activity. Microdeletions involving SHOX exons 1–6a and/or the CNEs result in idiopathic short stature (ISS) and Leri–Weill dyschondrosteosis (LWD). Here, we report six rare copy-number variations (CNVs) in PAR1 identified through copy-number analyzes of 245 ISS/LWD patients and 15 unaffected individuals. The six CNVs consisted of three microduplications encompassing SHOX and some of the CNEs, two microduplications in the SHOX 3′-region affecting one or four of the downstream CNEs, and a microdeletion involving SHOX exon 6b and its neighboring CNE. The amplified DNA fragments of two SHOX-containing duplications were detected at chromosomal regions adjacent to the original positions. The breakpoints of a SHOX-containing duplication resided within Alu repeats. A microduplication encompassing four downstream CNEs was identified in an unaffected father–daughter pair, whereas the other five CNVs were detected in ISS patients. These results suggest that microduplications involving SHOX cause ISS by disrupting the cis-regulatory machinery of this gene and that at least some of microduplications in PAR1 arise from Alu-mediated non-allelic homologous recombination. The pathogenicity of other rare PAR1-linked CNVs, such as CNE-containing microduplications and exon 6b-flanking microdeletions, merits further investigation.
Similar content being viewed by others

Introduction
SHOX located in the pseudoautosomal region 1 (PAR1) regulates skeletal development.1 SHOX haploinsufficiency underlies idiopathic short stature (ISS; MIM no. 300582), Leri–Weill dyschondrosteosis (LWD) characterized by Madelung deformity (MIM no. 249700) and less-specific skeletal changes including micrognathia.1, 2, 3, 4 SHOX transcripts include SHOXa encoded by exons 1–5 and 6a, and SHOXb encoded by exons 1–5 and 6b.1 SHOX expression requires multiple cis-acting elements in PAR1;5, 6, 7, 8, 9, 10, 11 previous studies have identified highly conserved non-coding DNA elements (CNEs) with cis-regulatory activity in the upstream (CNE-2, CNE-3 and CNE-5) and downstream regions (CNE4, CNE5, ECR1 and CNE9/ECS4) of SHOX (Supplementary Figure S1).7, 8, 10, 11
SHOX haploinsufficiency in cytogenetically normal individuals is mainly caused by microdeletions involving SHOX exons 1–6a and/or the CNEs.2, 3, 5, 6, 7, 8, 9 Recently, microduplications encompassing SHOX were also identified in patients with ISS and LWD.12, 13 However, the pathogenicity of these copy-number variations (CNVs) remains controversial, because SHOX overdosage has been associated with tall or normal stature.14, 15, 16, 17 Likewise, although a deletion in SHOX intron 6b was identified in an ISS family, its relevance to the disease phenotype remains unclear.18 Furthermore, the genomic origins of PAR1-linked CNVs have been poorly investigated.
Subjects and methods
Detailed methods are provided in the Supporting Information (Supplementary Methods). This study was approved by the Institutional Review Board Committee at the National Center for Child Health and Development and performed after obtaining informed consent. We searched for PAR1-linked CNVs in 245 patients with ISS or LWD and in 15 unaffected individuals. All patients had short stature with s.d. scores of <−2.0. Diagnosis of LWD was based on radiological findings of Madelung deformity.3 Individuals with chronic diseases or apparent chromosomal alterations were excluded. When possible, we analyzed participants’ parental samples. Genomic DNA samples were subjected to multiplex-ligation-dependent probe amplification. All CNVs, except for well-documented microdeletions involving SHOX and/or downstream CNEs,2, 3, 9 were further characterized by array-based comparative genomic hybridization. We also investigated the genomic structures of CNVs.
Results
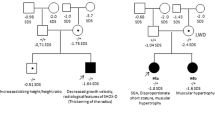
Rare CNVs were identified in patients 1–5 and control 1 (Figure 1,Table 1, and Supplementary Figure S2). Patients 1–3 carried microduplications involving SHOX and three or four of the seven CNEs. Patient 4 and control 1 had microduplications in the SHOX 3′-region encompassing one and all downstream CNEs, respectively. Patient 5 had a microdeletion involving exon 6b and CNE4. The CNV in patient 1 was a 270 737 bp tandem duplication, whose breakpoints resided within Alu repeats and shared a 12 bp overlap (Figure 2a). Fusion junctions of other CNVs could not be determined. Fluorescence in situ hybridization analysis of patient 2 using a SHOX-containing cosmid generated a signal only at Xp22.3 (Figure 2b), indicating that the amplified DNA fragment was probably inserted into a genomic interval adjacent to the original position. Patients 1–5 manifested mild or moderate short stature (Table 1). None of the five patients had Madelung deformity, whereas patient 3 showed micrognathia. The phenotypically normal father of control 1 had the same CNV as the proband.

(a)Array-based comparative genomic hybridization. The upper horizontal line indicates the physical distance from the Xp/Yp telomere (pter; hg19, Build 37). The purple box depicts SHOX. Orange and blue boxes denote highly evolutionarily conserved non-coding DNA elements (CNEs) in the SHOX upstream and downstream regions, respectively. The black, red and green dots denote normal, increased (log ratio⩾+0.5) and decreased (log ratio⩽–1.0) copy-numbers, respectively. The red and green arrows indicate duplicated and deleted regions, respectively. cen, centromere. (b) Schematic representation of the SHOX-flanking region. The upper horizontal line indicates the physical distance from the Xp/Yp telomere. The red and green lines denote the duplicated and deleted regions, respectively.

Characterization of copy-number variations (CNVs) in patients 1 and 2. (a) Upper panel: schematic representation of the duplication in patient 1. The purple boxes depict SHOX. Orange and blue boxes denote highly conserved non-coding DNA elements (CNEs) in the SHOX upstream and downstream regions, respectively. The red shaded area indicates the duplicated region. The genomic structure is not drawn to scale. Lower panel: DNA sequences at the fusion junction of the duplication in patient 1. The PCR product containing the junction was amplified using a primer pair, 5′-CCTCCAAAATAGCTGGCAATA-3′ and 5′-AGCATAAAATTCCCCATCTGA-3′. The CNV was a tandem duplication of 270 737 bp (chrX:486,700–757,437; hg19, Build 37). (b) Fluorescence in situ hybridization analysis of patient 2. The SHOX-containing cosmid probe (green) generated signals only at Xp22.3 (arrows). The red signals (arrowheads) indicate the control probe for the Xq/Yq telomere.
Discussion
This study provides further evidence that SHOX-containing microduplications account for a small fraction of the etiology of ISS. Our findings contradict prior observations that trisomy of PAR1 owing to chromosomal rearrangements leads to tall or normal stature.13, 14, 15, 16, 17 These conflicting data can be explained by assuming that relatively large duplications containing all SHOX exons and cis-acting enhancers lead to gene overexpression and resultant tall stature, whereas small duplications encompassing only a part of these components attenuate SHOX expression by disrupting the cis-regulatory machinery. Consistent with this, the microduplication in patient 1 increases the distance between SHOX exons and the CNEs, and the duplicated DNA fragment in patient 2 was detected at a genomic region adjacent to the original position. On the other hand, the results of patient 4 and control 1 question the pathogenicity of duplications affecting only the CNEs. To our knowledge, no previous study has shown that enhancer overdosage impairs gene expression. Nevertheless, PAR1 microduplications apart from SHOX may disrupt the cis-regulatory machinery. Indeed, CNE-containing microduplications were identified in multiple ISS patients.12, 19 Furthermore, the pathogenicity of the exon 6b-containing microdeletion in patient 5 remains unclear. SHOX exon 6b has not been implicated in skeletal development; however, elimination of SHOXb may affect the function of SHOXa isoform, because SHOXb dimerizes with SHOXa.20 Alternatively, the ISS of patient 5 may be ascribed to CNE4 deletion. Moreover, as a deletion in intron 6b has been identified in an ISS family,18 exon 6b-flainking regions may contain another enhancer.
Our data imply that the duplication in patient 1 arose from Alu-mediated non-allelic homologous recombination (NAHR). Because PAR1 is enriched with Alu repeats4 and Alu-mediated NAHR has been implicated in the development of a microdeletion involving SHOX,9 it is possible that NAHR underlies various PAR1-linked CNVs. However, other mechanisms such as non-homologous end-joining may also underlie these CNVs.
Patients 1–5 showed no signs of Madelung deformity, indicating that PAR1-linked duplications and exon 6b-containing deletions result in relatively mild phenotypes. However, the mild phenotypes in our patients may be related to their young ages, because skeletal features in patients with SHOX abnormalities usually ameliorate after puberty.3 Notably, patient 3 with a duplication encompassing SHOX exons 1–3 manifested micrognathia, which may be associated with SHOX dysfunction,2 whereas patients 1–2 with duplications of all SHOX exons manifested no skeletal deformities. These findings are consistent with previous findings that partial SHOX duplications exert more significant effects on skeletal development than complete duplications.12 Alternatively, partial SHOX duplications may have a broad phenotypic spectrum, because these CNVs were identified in several unaffected relatives of the patients.12
Collectively, our results indicate that SHOX-containing microduplications cause ISS by disrupting the cis-regulatory machinery of SHOX and that Alu-mediated NAHR underlies at least some of these microduplications. Further studies are necessary to clarify the pathogenicity of other rare PAR1-linked CNVs.
References
Rao, E., Weiss, B., Fukami, M., Rump, A., Niesler, B., Mertz, A. et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat. Genet. 16, 54–63 (1997).
Rappold, G., Blum, W. F., Shavrikova, E. P., Crowe, B. J., Roeth, R., Quigley, C. A. et al. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J. Med. Genet. 44, 306–313 (2007).
Binder, G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm. Res. Paediatr. 75, 81–89 (2011).
Blaschke, R. J. & Rappold, G. The pseudoautosomal regions, SHOX and disease. Curr. Opin. Genet. Dev. 16, 233–239 (2006).
Benito-Sanz, S., Thomas, N. S., Huber, C., Gorbenko del Blanco, D., Aza-Carmona, M., Crolla, J. A. et al. A novel class of pseudoautosomal region 1 deletions downstream of SHOX is associated with Leri-Weill dyschondrosteosis. Am. J. Hum. Genet. 77, 533–544 (2005).
Benito-Sanz, S., Aza-Carmona, M., Rodríguez-Estevez, A., Rica-Etxebarria, I., Gracia, R., Campos-Barros, A. et al. Identification of the first PAR1 deletion encompassing upstream SHOX enhancers in a family with idiopathic short stature. Eur. J. Hum. Genet. 20, 125–127 (2012).
Benito-Sanz, S., Royo, J. L., Barroso, E., Paumard-Hernández, B., Barreda-Bonis, A. C., Liu, P. et al. Identification of the first recurrent PAR1 deletion in Léri-Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer. J. Med. Genet. 49, 442–450 (2012).
Fukami, M., Kato, F., Tajima, T., Yokoya, S. & Ogata, T. Transactivation function of an approximately 800-bp evolutionarily conserved sequence at the SHOX 3' region: implication for the downstream enhancer. Am. J. Hum. Genet. 78, 167–170 (2006).
Fukami, M., Dateki, S., Kato, F., Hasegawa, Y., Mochizuki, H., Horikawa, R. et al. Identification and characterization of cryptic SHOX intragenic deletions in three Japanese patients with Léri-Weill dyschondrosteosis. J. Hum. Genet. 53, 454–459 (2008).
Chen, J., Wildhardt, G., Zhong, Z., Röth, R., Weiss, B., Steinberger, D. et al. Enhancer deletions of the SHOX gene as a frequent cause of short stature: the essential role of a 250kb downstream regulatory domain. J. Med. Genet. 46, 834–839 (2009).
Durand, C., Bangs, F., Signolet, J., Decker, E., Tickle, C. & Rappold, G. Enhancer elements upstream of the SHOX gene are active in the developing limb. Eur. J. Hum. Genet. 18, 527–532 (2010).
Benito-Sanz, S., Barroso, E., Heine-Suñer, D., Hisado-Oliva, A., Romanelli, V., Rosell, J. et al. Clinical and molecular evaluation of SHOX/PAR1 duplications in Leri-Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). J. Clin. Endocrinol. Metab. 96, E404–E412 (2011).
Thomas, N. S., Harvey, J. F., Bunyan, D. J., Rankin, J., Grigelioniene, G., Bruno, D. L. et al. Clinical and molecular characterization of duplications encompassing the human SHOX gene reveal a variable effect on stature. Am. J. Med. Genet. A. 149A, 1407–1414 (2009).
Iughetti, L., Capone, L., Elsedfy, H., Bertorelli, R., Predieri, B., Bruzzi, P. et al. Unexpected phenotype in a boy with trisomy of the SHOX gene. J. Pediatr. Endocrinol. Metab. 23, 159–169 (2010).
Ogata, T., Kosho, T., Wakui, K., Fukushima, Y., Yoshimoto, M. & Miharu, N. Short stature homeobox-containing gene duplication on the der(X) chromosome in a female with 45,X/46,X, der(X), gonadal dysgenesis, and tall stature. J. Clin. Endocrinol. Metab. 85, 2927–2930 (2000).
del Rey, G., Jasper, H., Bengolea, S. V., Boywitt, A., De Bellis, R. & Heinrich, J. J. Trisomy of the short stature homeobox-containing gene (SHOX) due to duplication/deletion of the X chomosome: clinical implications on the stature. Horm. Res. Paediatr. 74, 297–304 (2010).
Ottesen, A. M., Aksglaede, L., Garn, I., Tartaglia, N., Tassone, F., Gravholt, C. H. et al. Increased number of sex chromosomes affects height in a nonlinear fashion: a study of 305 patients with sex chromosome aneuploidy. Am. J. Med. Genet. A 152A, 1206–1212 (2010).
Sandoval, G. T., Jaimes, G. C., Barrios, M. C., Cespedes, C. & Velasco, H. M. SHOX gene and conserved noncoding element deletions/duplications in Colombian patients with idiopathic short stature. Mol. Genet. Genomic. Med. 2, 95–102 (2014).
Wit, J. M., van Duyvenvoorde, H. A., van Klinken, J. B., Caliebe, J., Bosch, C. A., Lui, J. C. et al. Copy number variants in short children born small for gestational age. Horm. Res. Paediatr. 82, 310–318 (2014).
Rao, E., Blaschke, R. J., Marchini, A., Niesler, B., Burnett, M. & Rappold, G. A. The Leri-Weill and Turner syndrome homeobox gene SHOX encodes a cell-type specific transcriptional activator. Hum. Mol. Genet. 10, 3083–3091 (2001).
Acknowledgements
This study was supported by JCR Pharmaceuticals.
DISCLAIMER
The sponsor had no role in the study design, in the collection, analysis or interpretation of data, in the writing of the report or in the decision to submit the report for publication.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
MF has received a research grant from JCR Pharmaceuticals. The remaining authors declare no conflict of interest.
Additional information
The Japanese SHOX study group M. Adachi (Kanagawa Children’s Medical Center), T. Tajima (Hokkaido University), T. Tanaka (Tanaka Growth Clinic), O. Arisaka and S. Koyama (Dokkyo Medical University), T. Hamajima (Aichi Children’s Health and Medical Center), O. Nose (Nose Clinic), K. Ozono and N. Namba (Osaka University), K. Nagasaki (Niigata University), T. Kamimaki (Shizuoka City Shimizu Hospital), S. Kanzaki (Tottori University), T. Ogata (Hamamatsu University School of Medicine), H. Tanaka (Okayama Saiseikai General Hospital), Y. Hasegawa (Tokyo Metropolitan Children’s Medical Center), K. Kobayashi (University of Yamanashi), S. Dateki (Nagasaki University), H. Mabe (Kumamoto University), I. Fujiwara (Tohoku University), S. Ida (Osaka Medical Center and Research Institute for Maternal and Child Health), T. Hasegawa (Keio University), A. Uematsu (Shizuoka Children’s Hospital), K. Kashimada (Tokyo Medical Dental University), K. Onigata (Shimane University), K. Miyako (Fukuoka Children’s Hospital), S. Yokoya and R. Horikawa (National Center for Child Health and Development) and M. Fukami (National Research Institute for Child Health and Development).
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Fukami, M., Naiki, Y., Muroya, K. et al. Rare pseudoautosomal copy-number variations involving SHOX and/or its flanking regions in individuals with and without short stature. J Hum Genet 60, 553–556 (2015). https://doi.org/10.1038/jhg.2015.53
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.53
This article is cited by
-
Rare dosage abnormalities flanking the SHOX gene
Egyptian Journal of Medical Human Genetics (2021)
-
Variants in the 5′UTR reduce SHOX expression and contribute to SHOX haploinsufficiency
European Journal of Human Genetics (2021)
-
Screening of the SHOX/PAR1 region using MLPA and miRNA expression profiling in a group of Egyptian children with non-syndromic short stature
Egyptian Journal of Medical Human Genetics (2020)
-
A 105 kb interstitial insertion in the Xq27.1 palindrome from pseudoautosomal region PAR1 causes a novel X-linked recessive compound phenotype
Journal of Translational Medicine (2019)
-
Unbalanced X;9 translocation in an infertile male with de novo duplication Xp22.31p22.33
Journal of Assisted Reproduction and Genetics (2019)
