
Abstract
Recent developments in molecular therapies for Duchenne muscular dystrophy (DMD) demand accurate genetic diagnosis, because therapies are mutation specific. The KUCG (Kobe University Clinical Genetics) database for DMD and Becker muscular dystrophy is a hospital-based database comprising 442 cases. Using a combination of complementary DNA (cDNA) and chromosome analysis in addition to conventional genomic DNA-based method, mutation detection was successfully accomplished in all cases, and the largest mutation database of Japanese dystrophinopathy was established. Among 442 cases, deletions and duplications encompassing one or more exons were identified in 270 (61%) and 38 (9%) cases, respectively. Nucleotide changes leading to nonsense mutations or disrupting a splice site were identified in 69 (16%) or 24 (5%) cases, respectively. Small deletion/insertion mutations were identified in 34 (8%) cases. Remarkably, two retrotransposon insertion events were also identified. Dystrophin cDNA analysis successfully revealed novel transcripts with a pseudoexon created by a single-nucleotide change deep within an intron in four cases. X-chromosome abnormalities were identified in two cases. The reading frame rule was upheld for 93% of deletion and 66% of duplication mutation cases. For the application of molecular therapies, induction of exon skipping was deemed the first priority for dystrophinopathy treatment. At one Japanese referral center, the hospital-based mutation database of the dystrophin gene was for the first time established with the highest levels of quality and patient's number.
Similar content being viewed by others


Introduction
Duchenne muscular dystrophy (DMD; MIM (Online Mendelian Inheritance in Man) no. 310200) and Becker muscular dystrophy (BMD; MIM no. 300376) are allelic X-linked recessive diseases caused by mutations in the dystrophin gene (MIM no. 300377). DMD is the most frequent inherited muscle disease affecting one in every 3500 male births. DMD is characterized by a rapidly progressive disease that is first recognized during childhood; affected individuals commonly lose their ability to walk before they turn 12 years old. In their third decade, they usually succumb because of either cardiac or respiratory failure. BMD has a slower rate of progression; affected individuals remain ambulatory beyond the age of 16 years and may lead near-normal lives. During the infantile period, DMD/BMD can be identified in boys by a high elevation of serum creatine kinase activity, even before muscle weakness manifests.1
The dystrophin gene located at Xp21.2 is characterized by its enormous size (2.4 Mb). It consists of 79 exons, forming a 14-kb mRNA transcript, and lengthy introns (up to 250 kb).2 Mutation studies on the dystrophin gene have focused on detecting deletions or duplications of one or more exons, and multiplex PCR that amplifies selected deletion-prone exons has been used as the most efficient method of mutation detection.3, 4 In these gross rearrangements, the reading frame rule explains the clinical difference between DMD and BMD at the molecular level; that is, deletions or duplications that shift the reading frame of the dystrophin mRNA (out-of-frame) lead to the more severe DMD phenotype, whereas the milder BMD phenotype occurs if the reading frame is preserved (in-frame).5 This rule has been applicable in >90% of cases.
The recent introduction of the quantitative PCR-based technique of multiplex ligation-dependent probe amplification (MLPA), which amplifies all 79 exons and is commercially available, has significantly improved mutation detection of deletions or duplications.6, 7, 8, 9 However, the identification of small mutations in the dystrophin gene remains challenging, because of the large number of exons and the huge size of the dystrophin gene. By combining quantitative PCR and direct sequencing technologies, the mutation detection rate has risen to 98% of DMD cases (106/108 total cases).10 In spite of technological advances in mutation analysis, a complete mutation spectrum has not been established in a large cohort, leaving some percentage of cases undiagnosed,11 and the dystrophin gene remain uncharacterized in some percentage of cases.12
Identifying a dystrophin mutation in all cases would permit a clinical diagnosis as the basis for proper genetic counseling, and is also a precondition for future molecular therapies, such as correction of the reading frame shift by induction of exon skipping, or suppression of nonsense mutation during translation.13, 14, 15 In Japan, however, there is no large-scale database for mutations of dystrophinopathy. Strategies for molecular therapies or for comprehensive molecular testing remain undefined.
In this study we described the complete mutation spectrum of 422 Japanese DMD/BMD cases and propose strategies for molecular therapies and genetic testing.
Materials and methods
Patients
Japanese DMD/BMD male patients from all of Japan (although mainly from the West) were referred to Kobe University Hospital, Kobe, Japan, for their molecular diagnosis. The KUCG (Kobe University Clinical Genetics) database for dystrophinopathy is the hospital based-database and was completed by identifying mutations in all dystrophinopathy case as of May 2009. This is the largest Japanese dystrophinopathy database, including 530 patients from 442 families. To abolish bias in counting the incidence of mutations, we only included one case per unrelated family in this report. Among 442 cases, 356 or 86 were clinically diagnosed with DMD or BMD, respectively, because of their symptoms or muscle biopsy findings. Extensive mutation analysis of the dystrophin gene was conducted on the supposition that a responsible mutation should be identified in every dystrophinopathy case. In some cases, muscle biopsy was conducted for immunohistochemical examination and dystrophin complementary DNA (cDNA) was analyzed as previously reported.16, 17, 18 Informed consent was obtained for molecular analysis and this study was approved by the ethics committees of Kobe University School of Medicine (approval no. 28 in 1998).
Methods
Our strategies for the detection of mutations in the dystrophin gene changed over time.16, 17, 19, 20 In brief, genomic DNA (gDNA) was first analyzed to detect deletion or duplication of one or more exons. If no responsible gross mutation was identified, muscle biopsies were obtained for dystrophin immunostaining to confirm the diagnosis.18 Dystrophin mRNA extracted from lymphocytes or muscle tissue was analyzed using reverse transcription PCR.16, 17
Analysis of gDNA
Peripheral blood samples were obtained from the patients and their family members. gDNA was isolated using standard phenol–chloroform extraction methods. At the beginning of this study, Southern blot analysis using dystrophin cDNA as a probe was performed with HindIII restriction enzyme-digested DNA as a template, as described previously.21, 22 Later, this time-consuming and laborious method was abandoned. In most of cases, multiplex PCR that amplified deletion-prone 19 exons was used for the first line of mutation detection,3, 4 followed by the second-line method of the Southern blotting.21 When no responsible gross mutation was identified, in some cases all 79 exons were PCR amplified from gDNA and subjected to direct sequencing as previously described.23, 24 In cases in which no responsible mutation could be detected, the dystrophin cDNA was analyzed.16 As a final step, chromosomal analysis was conducted (Mitsubishi Medience, Tokyo, Japan).
Currently, MLPA (SALSA MLPA KIT P034/035 DMD/Becker; MRC-Holland, Amsterdam, The Netherlands) is our first-line method to identify mutations in the dystrophin gene,20 because this allows detection of deletions or duplications in all 79 exons. This analysis was conducted by Mitsubishi Medience. In cases with loss of a single exon by MLPA, the exon structure was examined by another method before concluding that there is a single-exon deletion, because small mutations have been found to disturb MLPA amplification.20
Analysis of dystrophin mRNA
Reverse transcription PCR was used to analyze the dystrophin mRNA expressed in lymphocytes or skeletal muscle as previously described.16, 25 Full-length dystrophin cDNA was amplified as 10–20 separate, partially overlapping fragments and the amplified products were sequenced. When mutations were identified in cDNA, they were confirmed in gDNA by PCR amplification and direct sequencing of the corresponding genomic region. In cases with deep intron mutations, ambiguous insertions were identified in their cDNA sequence. Subsequently, genomic mutations leading to the pseudoexon inclusion were analyzed by direct sequencing of gDNA.
Sequencing of the amplified products
The amplified products were purified and sequenced either directly or after subcloning into a pT7 blue T vector (Novagen, Madison, WI, USA).23 The DNA sequence was determined using an automated DNA sequencer (model 373A; Applied Biosystems, Foster City, CA, USA).
Nucleotide numbers for all the mutations were designated according to the cDNA reference sequence in GenBank, accession number NM_004006.1 (RefSeq NM_004006.1), in which the ‘A’ of the start codon is nucleotide 1.
Genotype–phenotype correlation
Because the reading frame rule is thought to explain the phenotype,5 this was examined in all cases. When the exact size of the deletion or duplication was known, the resulting reading frame was designated as out-of-frame or in-frame. For phenotype classification, DMD was diagnosed when cases became wheelchair-bound by the age of 12 years or when dystrophin deficiency was detected on muscle biopsy. BMD was diagnosed when cases could walk unsupported above the age of 13 years.
Results
In this study, all dystrophinopathy cases referred to the Kobe University Hospital for mutation analysis of the dystrophin gene were shown to have a responsible mutation, establishing the KUCG database. Mutations were identified in 442 DMD and BMD cases (Figure 1a), which included 260 different mutations.

(a–c) Full spectrum of mutations identified in the dystrophin gene among Japanese dystrophinopathy cases. (a) Distribution of mutations in 442 dystrophinopathy cases. Gross changes in gDNA were detected by PCR, Southern blotting or multiplex ligation-dependent probe amplification (MLPA) analysis. After this step, deletions and duplications encompassing one or more exons were identified in 270 (61%) and 38 cases (9%), respectively. For the second-line analysis, dystrophin cDNA or exons and intron/exon boundaries of gDNA were sequenced. Nonsense mutations were found in 69 cases (16%). Splice site mutations and small insertion/deletion mutations from 1 to 606 bp long were identified in 24 (5%) and 34 cases (8%), respectively. In four cases (1%), dystrophin cDNA analysis identified novel transcripts with a pseudoexon created by a single nucleotide change deep into an intron. X-chromosome abnormalities were identified in two cases (0.5%). Distribution of mutations in 356 DMD cases (b) and 86 BMD cases (c).
Among the 442 mutation events, 270 (61%) and 38 (9%) were large deletions or duplications of one or more exons, respectively. In other words, large deletions and duplications were observed in 308 cases (70%). The ratio of duplication to deletion was 0.14 (38/270), with deletion being seven times more common than duplication. This indicates that deletion and duplication are not reciprocal events. Nonsense mutations were identified in 69 cases (16%). Mutations disrupting the splice site consensus sequences were detected in 24 cases (5%). Small deletions/insertions (one to several hundreds nucleotides) were identified in 34 cases (8%). Interestingly, four deep intron mutations that created pseudoexons with novel splicing consensus sequences were identified in four cases (1%) using dystrophin cDNA analysis. Chromosomal abnormalities were detected in two cases (0.5%). In one BMD case, a splicing abnormality was identified, although no responsible genomic change was evident.26 It seems that the frequency of these mutations differed depending on the phenotypic group, as shown in Figures 1b and c. A high frequency (67%) of large deletion is observed in BMD cases when compared with DMD cases (60%), whereas nonsense mutations represent only 3% of the mutations in BMD cases when compared with 19% in DMD cases.
Large deletions and duplications
Large deletions of one or more exons were identified in 270 cases (Figure 1). The deletion patterns were categorized into 104 patterns (Figures 2a and b), in which 5 patterns were observed in both DMD and BMD (deletions of exons 3–7, 10–44, 43, 44 and 45). The most common pattern was in-frame deletion of exons 45–47, which was found in 12 cases (4.4%); all were BMD (Figure 2b). The second most common pattern was out-of-frame deletion of exons 45–52, 48–50 or the single exon 52; each was found in 11 cases and all of these were DMD (Figure 2a). The largest deletion encompassed exons 1–59, and was found in one case (KUCG no. 513). A single-exon deletion was the most frequently observed pattern, representing 19 patterns among 65 cases. After exon 52 deletion, the second most common single-exon deletion was of exons 44 or 45, both of which were identified in 10 cases (9 DMD and 1 BMD cases with exon 44 deletion, and 8 DMD and 2 BMD cases with exon 45 deletion). Deletions were found to cluster in proximal or distal hotspots (Figure 2). Deletions starting in the distal hotspot (exons 45–55) represented 65% (175/270) of the identified deletions, whereas deletions starting in the proximal hotspot (exons 2–20) accounted for only 26% (69/270). Intron 44 was by far the most frequently involved in deletion breakpoints, preferentially as a starting breakpoint.

Patterns of exon deletion in DMD (a) and BMD cases (b). Exon deletions were categorized into 104 patterns. Deleted exon regions are represented by horizontal bars; filled or shaded bars represent out-of-frame or in-frame deletions, respectively. On the bottom line, exons are numbered 1–79. The number at the end of each bar represents the number of cases with an identical deletion, whereas no number means that the deletion is unique. Asterisks indicate the deletion mutations that could result in both DMD and BMD phenotypes. Deletions were clustered in two hotspots: nearly 1/3 were in the proximal region of the gene extending from exons 2 to 20 and the other 2/3 were in the central region extending from exons 45 to 55 in both DMD and BMD cases.
Large duplications were identified in 38 cases (9%) and their duplication patterns were categorized into 31 patterns, in which duplication of exon 2 was observed in both DMD and BMD (Figures 3a and b), indicating considerable heterogeneity among the duplication mutations. Out of 31 patterns, 27 were observed only once, whereas 4 were repeated duplication events. Duplication of exon 2 was the most common, observed in four cases (two DMD and two BMD cases). The second most common duplication was that of exons 3–7, found in three DMD cases. Single-exon duplication was observed in six patterns and the largest duplication stretched from exons 3–43. It is noteworthy that the duplicated regions were not in tandem but separated into two regions in two different patterns (the ninth and eleventh lines in Figure 3a).27 The duplicated exons were distributed across the dystrophin gene and the proximal hotspot was more frequently duplicated than the distal hotspot with deletion mutations (Figures 2 and 3).

Patterns of exon duplication in DMD (a) and BMD cases (b). Exon duplications were categorized into 31 patterns from 38 cases. Bars represent the location of duplicated exons and filled and shaded bars represent out-of-frame or in-frame duplications, respectively. On the ninth and eleventh lines from the top in a, dotted lines indicate normal exons between separated duplications (duplication of exons 45–48 and 55–63, and exons 19–31 and 45–51, respectively). The number at the end of each line represents the number of cases with an identical duplication, whereas no number means that the duplication is unique. Asterisks indicate the duplication mutations that could result in both DMD and BMD phenotypes.
Analysis of the breakpoint distribution pattern in the dystrophin gene identified differences between the deletion and duplication mutation events (Figures 2 and 3). Deletion breakpoints were clustered in a few introns in the distal deletion hotspot, whereas they were more disseminated in the proximal region in accordance with the observed higher diversity among duplication events. Exon deletions involved multiple exons with a mean and median of 5.9 and 4 exons, respectively, whereas those for exon duplication were 10.0 and 5 exons, respectively. A greater variability in deletion length was associated with the proximal hotspot, in which more than 10 exons were frequently deleted.
Nonsense mutations
Nonsense mutations represented 16% of all the mutations (69/442) and were the second most common category (Figure 1a). These 62 patterns of single-nucleotide changes were dispersed across the dystrophin gene (Table 1), and 18 were novel (26%). Most were unique but c.5899C>T was found in three cases, and c.2365G>T, c.4729C>T, c.8608C>T, c.9568C>T and c.10108C>T were found in two cases each (Table 1). Among these six repeated nonsense mutations, five, including c.5899C>T, were CGA to TGA mutations. In fact, CGA to TGA transitions comprised nearly a quarter of nonsense mutations (18/69 cases; Table 1). This is because the CpG site is vulnerable to deamination, in which C is replaced by T.28, 29 The CGA codon is found 29 times in the dystrophin gene, and 12 of them (41%) were found to be mutated to TGA in this study. Although nonsense mutations usually cause DMD, three nonsense mutations were identified in the milder, BMD cases (4%) (Table 1). Two were found to produce in-frame dystrophin mRNA with skipping of the nonsense-containing exon,17, 30 and the other was present in the C-terminal region.31
Deep intron mutations
In all, four deep intron mutations, located at least 285 bp from an intron/exon junction, were identified (Figure 4). These would not have been identified by conventional gDNA analysis that merely analyzes intron/exon boundaries. In these cases, reverse transcription PCR of dystrophin mRNA showed larger amplified products than expected, and sequencing of the products revealed that additional exonic sequence that matched part of an intron was inserted between authentic exons (Figure 4). In one BMD case (KUCG no. 9597), a thymine 5590 bp into intron 2 was replaced with an adenine. This resulted in a splice acceptor consensus sequence and a novel 132-bp exon was included between exons 2 and 332 (Figure 4a). In another DMD case (KUCG no. 465), a cytosine 843 bp upstream of exon 28 was replaced with an adenine, producing the consensus splice donor site. Therefore, a novel 119-bp exon was created and incorporated into the dystrophin mRNA (Figure 4b). The other two cases (KUCG nos. 290 and 857) also had single-nucleotide changes deep into an intron 285 or 2714 bp from an intron/exon boundary, respectively, which created splice donor sites. In these cases, pseudoexons (58 and 121 bp long, respectively) were inserted between authentic exons (Figures 4c and d). Insertions of out-of-frame pseudoexons resulted in DMD in KUCG no. 857. However, KUCG no. 290, who also had an insertion of an out-of-frame pseudoexon, showed a BMD phenotype because mRNA without the pseudoexon was also transcribed.

Deep intron mutations. Four deep intron mutations are schematically described. Boxes and lines represent exons and introns, respectively. The shaded boxes represent pseudoexons. Numbers in the open boxes indicate the exon number of the dystrophin gene, and the size of the pseudoexon is indicated above the shaded box. Two to five nucleotides adjacent to the pseudoexons are shown on the intron lines, and the mutated nucleotides are shown by vertical arrows. The upper and lower horizontal square brackets indicate distance from the intron/exon border and intron size, respectively. (a) In a BMD case (KUCG no. 9597), a novel splice site was created and a 132-bp exon was created and incorporated into the mRNA. (b) In a DMD case (KUCG no. 465), a novel splice site was created and a 119-bp exon was created and incorporated into the mRNA. (c) In a BMD case (KUCG no. 290), a novel splice site was created, and a 58-bp exon was created and incorporated into the mRNA. (d) In a DMD case (KUCG no. 857), a novel splice site was created, and a 121-bp exon was created and incorporated into the mRNA.
Splice site mutations
Nucleotide changes disrupting the splice site consensus sequence were shown to produce abnormal transcripts in 24 cases (Table 2). Among 24 splice site mutations, 22 were single-nucleotide changes and the other two were a 2-bp insertion and 8-bp deletion, each found in one case. The most common nucleotide change was a G-to-A change at the first intronic nucleotide; this was found in different introns of five cases. A nucleotide change at the first or last nucleotide of an intron was identified in 11 cases, accounting for 46% of all splice site mutations. Mutations at the splice donor site were more common than those at the splice acceptor site (17 vs 7 cases). In this study, no mutations were identified at the branch point. The splicing pathway affected by these mutations differed from mutation to mutation.33 Exon skipping was occurring in 12 cases. Cryptic splice site activation occurred in 12 cases, and in 3 cases both exon skipping and cryptic splice site activation occurred.
Other mutations
Thirty four small mutations, ranging from 1 to 606 bp, were identified (Table 3) and all were unique. Their locations were dispersed and no clustering was identified. Deletions ranging from 1 to 52 bp were identified in 25 cases. Insertions ranging from 1 to 606 bp were identified in nine cases. Two insertions, which were 606 bp within exon 44 and 325 bp within exon 67, respectively, were found to be identical in DNA sequence to retrotransposons.34
In one BMD case a splicing error was detected, although there was no mutation in the gDNA (KUCG no. 338 in Table 4).26 Two previously unreported chromosomal abnormalities were found, once each (Table 4).
The frequency of small mutation identification was examined in all exons and their neighboring introns. Small mutations were identified six times in exon 44, and five times each in exons 8, 34 and 70. The length of exons 8, 34, 44 and 70 was 182, 171, 148 and 137 bp, respectively; that is, both longer and shorter than the mean dystrophin exon length of 143 bp, and hence the high mutation frequency in these exons does not reflect exon length. This suggests an unknown mutagenic susceptibility within these exons.
Genotype–phenotype correlations
The reading frame rule was examined in these deletion cases (Figure 2). Out of 270 cases, 252 complied with the reading frame rule (93.3%), whereas 18 cases (6.7%) did not: 9 DMD cases possessed an in-frame deletion (Figure 2a) and 9 BMD cases showed an out-of-frame deletion (Figure 2b). The exception to the rule was observed more frequently in BMD (15.5%) than in DMD (4.2%). It is remarkable that identical deletions could result in two different phenotypes; 3 out of 9 cases with exons 3–7 deletions, 2 out of 3 cases with exon 43 deletions, 1 out of 10 cases with exon 44 deletions and 2 out of 10 cases with exon 45 deletions show mild phenotype in spite of out-of-frame deletion, and 1 out of 2 cases with exons 10–44 deletions show severe phenotype in spite of in-frame deletion.
Among duplication mutations, 25 complied with the reading frame rule (65.8%) but 13 did not (34.2%): 7 DMD cases showed an in-frame duplication (Figure 3a) and 6 BMD cases had an out-of-frame duplication (Figure 3b). The incidence of cases that did not fit with the reading frame rule was higher among duplications (34.2%) than among the deletion mutations (6.7%). Dual phenotypes with an identical duplication were observed for exon 2 duplication.
Discussion
In this study, pathogenic mutations in the dystrophin gene were successfully identified in all 442 Japanese DMD/BMD cases (Figure 1), and 49 (39%) of the identified small mutations were novel (Tables 1, 2 and 3). This has enabled all Japanese dystrophinopathy cases to receive a proper diagnosis or genetic counseling and will also expedite the application of mutation-specific molecular therapies. The KUCG database is one of the largest mutation databases constructed at a single institute and is characterized by its high quality, because all the dystrophin mutations were identified and confirmed within one institute under a single principal investigator, and the clinical data from all patients are regularly updated at follow-up visits to the outpatient clinic.
Several mutation studies analyzing more than 100 DMD/BMD cases have been reported in the literature. The largest institutional database reported was from Italy; that database contained 506 cases, but some cases remained for which no dystrophin mutation had been identified.35 The KUCG database is the second-largest institutional database, but the first completed mutation database. Even in small-scale studies, the mutation detection rate has previously only reached up to 96% of examined cases.10, 11, 36, 37 It was believed that a causative mutation in the dystrophin gene could not be identified in approximately 1 to 2% of DMD/BMD patients.12 In contrast to previous studies, our perfect mutation detection rate became possible by conducting dystrophin cDNA or chromosomal analysis. Without these analyses, our mutation detection rate decreased by nearly 2%, that is, down to the previously accepted detection rate.12 We propose that similarly extensive mutation analysis should be performed to identify the responsible mutation when dystrophin deficiency has been confirmed.
An existing locus-specific database for dystrophinopathy in Leiden focuses on mutations that have been either reported in the literature or directly submitted to the database.38 Because of the involvement of multiple submitters, the data quality is not homogeneous and analyses could be biased. The French database (UMD-DMD France), containing 2084 independent mutations in dystrophin, has been established by collaboration with several institutes, and the quality of UMD-DMD France is guaranteed by curators.29
For a long time, multiplex PCR examining selected deletion-prone exons has been used as the mutation screening method of choice for the dystrophin gene. Our results indicate that using MLPA to examine all 79 exons for deletion or duplication mutations allows mutation detection in 70% of Japanese DMD/BMD cases (Figure 1). In addition, MLPA has been shown to detect small mutations.20 MLPA is considered to be the most powerful single technology to identify mutations in the huge dystrophin gene, and should be used as the first-line mutation detection method for the dystrophin gene in Japanese dystrophinopathy. In contrast, in Taiwan, which is geographically near to Japan, MLPA analysis identified a lower incidence of deletions and a higher incidence of duplications (deletion and duplication mutations were found in 36.0 and 24.7% of 89 DMD/BMD patients, respectively39), compared with other reports that show 60% and 5–10%, respectively.6, 8, 40 Although differences in ethnicity should be taken into account, MLPA remains the first line in mutation detection in dystrophinopathy.6, 7, 8, 9
A total of four deep intron mutations were identified at least 285 bp from the exon/intron boundary; this was possible only because we used detection of novel dystrophin mRNA (Figure 4). This analysis provided 1% of detected mutations, and facilitated our perfect mutation detection rate. Therefore, we suggest that it is necessary to analyze dystrophin mRNA to complete the mutation analysis, even when the dystrophin gene is apparently normal. Deep intron mutations have previously been identified in three cases, and their potential to be treated by exon skipping strategies was examined. Because the completely normal 79 exons of the dystrophin gene were maintained in these cases, induction of pseudoexon skipping would be expected to result in completely normal expression of dystrophin, the ultimate goal of dystrophinopathy treatment.41 Our cases can now be tested for any potential to be treated by exon skipping.
In contrast to the deletions and duplications, which were localized within two hotspots, nonsense mutations were detected throughout the gene, with 30.4% (21), 30.4% (21), 24.6% (17) and 14.5% (10) in the first (exons 1–20), second (exons 21–40), third (exons 41–60) and fourth (exons 61–79) quartiles of the dystrophin gene, respectively. Although nonsense mutations were detected in 16% of all cases in Japan, reports from outside Japan have described lower nonsense mutation rates up to 13.2%.10, 29, 42 The rate of nonsense mutation is high among the Japanese. This may be because of a difference in the effort expended in finding nonsense mutations in these reports.
It has been reported that one nonsense mutation at a CpG site in exon 59 (c.8713C>T; p.R2905X) was detected in six patients with different haplotypes,28 and this mutation has been reported from several countries, suggesting that c.8713C>T is a hotspot for mutation. In this study, however, no case showed this point mutation. In contrast, in the Japanese, the most common nucleotide change was c.5899C>T, which was found in three cases (Table 1). Interestingly, c.5899C>T has been once detected in the United States.28 In Taiwan, which is geographically near to Japan, c.10108C>T was identified three times among 14 nonsense mutation cases,42 and this mutation was identified twice in our study. These findings suggest an ethnic difference in the mutability of CpG sites, involving the rate of demethylation or features of the chromatin structure.
Although the enormous dystrophin gene has huge introns that have accumulated retrotransposon insertions, this has rarely been shown to cause disease.43 In our series, two retrotransposon insertions were found.34 Their identification was because of the elongation of exon size, which made PCR amplification difficult, and could have resulted in misdiagnosis as a single-exon deletion. Therefore, we propose that supposed single-exon deletion cases should be examined in more detail.
Although the reading frame rule is thought to explain the phenotype,5 18 out of 270 cases with deletion (6.7%) and 13 out of 38 cases with duplication (34.2%) did not fit with the reading frame rule (Figures 2 and 3). Furthermore, identical mutations could result in two different phenotypes; deletions of exons 3–7, 10-44, 43, 44 and 45, and duplication of exon 2. In-frame deletions starting in the actin-binding domain or disrupting the cysteine-rich domain resulted in DMD phenotype because of the lack of binding sites with dystrophin-associated proteins. Other mechanisms may have a role in modulating the clinical severity, including recoding mechanism and unusual splicing.29
We examined the applicability for molecular therapies among the 260 different mutations identified in DMD/BMD (Table 5). Nonsense suppression therapy has been studied and a compound named PTC124 is now in clinical trials to treat DMD cases caused by a nonsense mutation.15 This treatment could be applied to 66 cases with nonsense mutations in our study (19% of DMD cases). Induction of exon skipping that corrects out-of-frame to in-frame has been proposed as a highly plausible method for DMD treatment, and the first clinical trial has been conducted in Japan.13, 44 The applicability of this technology was examined in the KUCG database. The greatest number of treatable cases with one antisense oligonucleotide was 40, which could be treated by skipping of the single exon 51. In fact, exon 51 skipping treatment has already been clinically tested in Europe.14 Skipping of exon 53 would produce an in-frame transcript in 39 DMD cases. In total, single-exon skipping could be applied to the treatment of 173 cases with 58 patterns of deletion. Skipping of two or three exons could be applied to 27 and 1 case, respectively. It seems therefore that induction of exon skipping is the most applicable molecular therapy for the treatment of DMD. However, it is noteworthy that dual phenotypes can result from one identical deletion (Figures 2a and b), and this must be considered when assessing the potential effects of the treatment. With exon skipping treatment, the most effective results would be expected for the four deep intron mutations (Figure 4), because expression of a completely normal dystrophin transcript could be induced.
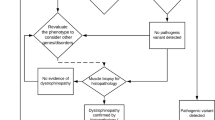
The best strategy for comprehensive dystrophin gene testing has long been debated. A balance must be found between the need to obtain definitive results and the effort required to perform a comprehensive analysis of heterogeneous mutations in the large dystrophin gene.10 Recently, one analytical method has been shown to identify mutations in the dystrophin gene in 98% of DMD cases.37 This is very useful but is still not perfect in identifying all mutations. From our results, it is possible to define a strategy that will be useful in prioritizing cases for various stages of dystrophin gene mutation analysis and will maximize the benefits of comprehensive mutation analysis for dystrophinopathy (Figure 5). In this strategy, MLPA analysis should be used first, to detect 70% of the mutations (step 1). Second, direct sequencing of the four exons 8, 34, 44 and 70 (which each contained small mutations in five or six cases; Tables 1, 2 and 3) should be conducted to reveal mutations in a further 4.5% of cases (step 2). Thereafter, muscle biopsy should be conducted to confirm the diagnosis and obtain dystrophin mRNA for sequence analysis. Dystrophin cDNA analysis is expected to reveal mutations in 25% of cases (step 3). Finally, chromosome analysis would complete the mutation analysis (step 4), enabling mutation detection in all dystrophinopathy cases.

Molecular testing strategy for Japanese dystrophinopathy. A diagram for the molecular diagnosis of dystrophinopathy (a) and the frequency of mutation detection by each diagnostic step (b). Multiplex ligation-dependent probe amplification (MLPA) analysis identifies deletion and duplication mutations encompassing one or more exons, which account for 70% of dystrophinopathy cases. In cases with loss of a single exon by MLPA, the exon structure was examined by another method before concluding that there is a single-exon deletion, because small mutations have been found to disturb MLPA amplification.20 Sequencing of exons and flanking introns of exons 8, 34, 44 and 74, in which small mutations are detected frequently, revealed mutations in a further 4.5% of dystrophinopathy cases. After confirmation of the diagnosis as dystrophinopathy, cDNA analysis was performed, which identified mutations in 25% of cases. After chromosome analysis, mutations were identified in all dystrophinopathy cases.
Accession codes
References
Zatz, M., Rapaport, D., Vainzof, M., Passos-Bueno, M. R., Bortolini, E. R., Pavanello, R. C. M. et al. Serum creatine-kinase (CK) and pyruvate-kinase activities in Duchenne (DMD) as compared with Becker (BMD) muscular dystrophy. J. Neurol. Sci. 102, 190–196 (1991).
Ahn, A. H. & Kunkel, L. M. The structural and functional diversity of dystrophin. Nat. Genet. 3, 283–291 (1993).
Chamberlain, J. S., Gibbs, R. A., Ranier, J. E., Nguyen, P. N. & Caskey, C. T. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 16, 11141–11156 (1988).
Beggs, A. H., Koenig, M., Boyce, F. M. & Kunkel, L. M. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum. Genet. 86, 45–48 (1990).
Monaco, A. P., Bertelson, C. J., Liechti-Gallati, S., Moser, H. & Kunkel, L. M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 2, 90–95 (1988).
White, S., Kalf, M., Liu, Q., Villerius, M., Engelsma, D., Kriek, M. et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am. J. Hum. Genet. 71, 365–374 (2002).
Gatta, V., Scarciolla, O., Gaspari, A. R., Palka, C., De Angelis, M. V., Di Muzio, A. et al. Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA). Hum. Genet. 117, 92–98 (2005).
Lalic, T., Vossen, R., Coffa, J., Schouten, J., Guc-Scekic, M., Radivojevic, D. et al. Deletion and duplication screening in the DMD gene using MLPA. Eur. J. Hum. Genet. 13, 1231–1234 (2005).
Janssen, B., Hartmann, C., Scholz, V., Jauch, A. & Zschocke, J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: potential and pitfalls. Neurogenetics 6, 29–35 (2005).
Stockley, T. L., Akber, S., Bulgin, N. & Ray, P. N. Strategy for comprehensive molecular testing for Duchenne and Becker muscular dystrophies. Genet. Test. 10, 229–243 (2006).
Zeng, F., Ren, Z., Huang, S., Kalf, M., Mommersteeg, M., Smit, M. et al. Array-MLPA: comprehensive detection of deletions and duplications and its application to DMD patients. Hum. Mutat. 29, 190–197 (2008).
Hegde, M. R., Chin, E. L., Mulle, J. G., Okou, D. T., Warren, S. T. & Zwick, M. E. Microarray-based mutation detection in the dystrophin gene. Hum. Mutat. 29, 1091–1099 (2008).
Takeshima, Y., Yagi, M., Wada, H., Ishibashi, K., Nishiyama, A., Kakumoto, M. et al. Intravenous infusion of an antisense oligonucleotide results in exon skipping in muscle dystrophin mRNA of Duchenne muscular dystrophy. Pediatr. Res. 59, 690–694 (2006).
van Deutekom, J., Janson, A., Ginjaar, I., Frankhuizen, W., Aartsma-Rus, A., Bremmer-Bout, M. et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 357, 2677–2686 (2007).
Welch, E. M., Barton, E. R., Zhuo, J., Tomizawa, Y., Friesen, W. J., Trifills, P. et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 447, 87–91 (2007).
Matsuo, M., Masumura, T., Nishio, H., Nakajima, T., Kitoh, Y., Takumi, T. et al. Exon skipping during splicing of dystrophin mRNA precursor due to an intraexon deletion in the dystrophin gene of Duchenne muscular dystrophy Kobe. J. Clin. Invest. 87, 2127–2131 (1991).
Shiga, N., Takeshima, Y., Sakamoto, H., Inoue, K., Yokota, Y., Yokoyama, M. et al. Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible for Becker muscular dystrophy. J. Clin. Invest. 100, 2204–2210 (1997).
Adachi, K., Takeshima, Y., Wada, H., Yagi, M., Nakamura, H. & Matsuo, M. Heterogous dystrophin mRNAs produced by a novel splice acceptor site mutation in intermediate dystrophinopathy. Pediatr. Res. 53, 125–131 (2003).
Matsuo, M., Masumura, T., Nakajima, T., Kitoh, Y., Takumi, T., Nishio, H. et al. A very small frame-shifting deletion within exon 19 of the Duchenne muscular dystrophy gene. Biochem. Biophys. Res. Commun. 170, 963–967 (1990).
Okizuka, Y., Takeshima, Y., Awano, H., Zhang, Z., Yagi, M. & Matsuo, M. Small mutations detected by multiplex ligation-dependant probe amplification of the dystrophin gene. Genet. Test Mol. Biomarkers 13, 427–431 (2009).
Patria, S. Y., Takeshima, Y., Suminaga, R., Nakamura, H., Iwasaki, R., Minagawa, T. et al. A simple explanation for a case of incompatibility with the reading frame theory in Duchenne muscular dystrophy: failure to detect an aberrant restriction fragment in Southern blot analysis. Brain Dev. 21, 386–389 (1999).
Koenig, M., Hoffman, E. P., Bertelson, C. J., Monaco, A. P., Feener, C. & Kunkel, L. M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50, 509–517 (1987).
Tran, V. K., Takeshima, Y., Zhang, Z., Yagi, M., Nishiyama, A., Habara, Y. et al. Splicing analysis disclosed a determinant single nucleotide for exon skipping caused by a novel intra-exonic four-nucleotide deletion in the dystrophin gene. J. Med. Genet. 43, 924–930 (2006).
Tran, V. K., Takeshima, Y., Zhang, Z., Habara, Y., Haginoya, K., Nishiyama, A. et al. A nonsense mutation-created intraexonic splice site is active in the lymphocytes, but not in the skeletal muscle of a DMD patient. Hum. Genet. 120, 737–742 (2007).
Roberts, R. G., Barby, T. F., Manners, E., Bobrow, M. & Bentley, D. R. Direct detection of dystrophin gene rearrangements by analysis of dystrophin mRNA in peripheral blood lymphocytes. Am. J. Hum. Genet. 49, 298–310 (1991).
Patria, S. Y., Alimsardjono, H., Nishio, H., Takeshima, Y., Nakamura, H. & Matsuo, M. A case of Becker muscular dystrophy resulting from the skipping of four contiguous exons (71-74) of the dystrophin gene during mRNA maturation. Proc. Assoc. Am. Phys. 108, 308–314 (1996).
Zhang, Z., Takeshima, Y., Awano, H., Nishiyama, A., Okizuka, Y., Yagi, M. et al. Tandem duplications of two separate fragments of the dystrophin gene in a patient with Duchenne muscular dystrophy. J. Hum. Genet. 53, 215–219 (2008).
Buzin, C. H., Feng, J., Yan, J., Scaringe, W., Liu, Q., den Dunnen, J. et al. Mutation rates in the dystrophin gene: a hotspot of mutation at a CpG dinucleotide. Hum. Mutat. 25, 177–188 (2005).
Tuffery-Giraud, S., Beroud, C., Leturcq, F., Yaou, R. B., Hamroun, D., Michel-Calemard, L. et al. Genotype-phenotype analysis in 2405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum. Mut. 30, 934–945 (2009).
Nishiyama, A., Takeshima, Y., Zhang, Z., Habara, Y., Tran, T. H., Yagi, M. et al. Dystrophin nonsense mutations can generate alternative rescue transcripts in lymphocytes. Ann. Hum. Genet. 72, 717–724 (2008).
Suminaga, R., Takeshima, Y., Wada, H., Yagi, M. & Matsuo, M. C-terminal truncated dystrophin identified in skeletal muscle of an asymptomatic boy with a novel nonsense mutation of the dystrophin gene. Pediatr. Res. 56, 739–743 (2004).
Yagi, M., Takeshima, Y., Wada, H., Nakamura, H. & Matsuo, M. Two alternative exons can result from activation of the cryptic splice acceptor site deep within intron 2 of the dystrophin gene in a patient with as yet asymptomatic dystrophinopathy. Hum. Genet. 112, 164–170 (2003).
Habara, Y., Takeshima, Y., Awano, H., Okizuka, Y., Zhang, Z., Saiki, K. et al. In vitro splicing analysis reveals that availability of a cryptic splice site is not a determinant for alternative splicing patterns caused by +1G>A mutations in introns of the dystrophin gene. J. Med. Genet. 46, 542–547 (2009).
Narita, N., Nishio, H., Kitoh, Y., Ishikawa, Y., Ishikawa, Y., Minami, R. et al. Insertion of a 5′ truncated L1 element into the 3′ end of exon 44 of the dystrophin gene resulted in skipping of the exon during splicing in a case of Duchenne muscular dystrophy. J. Clin. Invest. 91, 1862–1867 (1993).
Trimarco, A., Torella, A., Piluso, G., Maria Ventriglia, V., Politano, L. & Nigro, V. Log-PCR: a new tool for immediate and cost-effective diagnosis of up to 85% of dystrophin gene mutations. Clin. Chem. 54, 973–981 (2008).
Taylor, P. J., Maroulis, S., Mullan, G. L., Pedersen, R. L., Baumli, A., Elakis, G. et al. Measurement of the clinical utility of a combined mutation detection protocol in carriers of Duchenne and Becker muscular dystrophy. J. Med. Genet. 44, 368–372 (2007).
Ashton, E. J., Yau, S. C., Deans, Z. C. & Abbs, S. J. Simultaneous mutation scanning for gross deletions, duplications and point mutations in the DMD gene. Eur. J. Hum. Genet. 16, 53–61 (2008).
Aartsma-Rus, A., Van Deutekom, J. C., Fokkema, I. F., Van Ommen, G. J. & Den Dunnen, J. T. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 34, 135–144 (2006).
Hwa, H. L., Chang, Y. Y., Chen, C. H., Kao, Y. S., Jong, Y. J., Chao, M. C. et al. Multiplex ligation-dependent probe amplification identification of deletions and duplications of the Duchenne muscular dystrophy gene in Taiwanese subjects. J. Formos. Med. Assoc. 106, 339–346 (2007).
Schwartz, M. & Duno, M. Improved molecular diagnosis of dystrophin gene mutations using the multiplex ligation-dependent probe amplification method. Genet. Test. 8, 361–367 (2004).
Gurvich, O. L., Tuohy, T. M., Howard, M. T., Finkel, R. S., Medne, L., Anderson, C. B. et al. DMD pseudoexon mutations: splicing efficiency, phenotype, and potential therapy. Ann. Neurol. 63, 81–89 (2008).
Hwa, H. L., Chang, Y. Y., Huang, C. H., Chen, C. H., Kao, Y. S., Jong, Y. J. et al. Small mutations of the DMD gene in Taiwanese families. J. Formos. Med. Assoc. 107, 463–469 (2008).
Musova, Z., Hedvicakova, P., Mohrmann, M., Tesarova, M., Krepelova, A., Zeman, J. et al. A novel insertion of a rearranged L1 element in exon 44 of the dystrophin gene: further evidence for possible bias in retroposon integration. Biochem. Biophys. Res. Commun. 347, 145–149 (2006).
Matsuo, M. Duchenne/Becker muscular dystrophy: from molecular diagnosis to gene therapy. Brain Dev. 18, 167–172 (1996).
Acknowledgements
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan, a Health and Labor Science Research Grant (Research on Psychiatric and Neurological Diseases and Mental Health) and a Research Grant for Nervous and Mental Disorders from the Ministry of Health, Labor and Welfare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Takeshima, Y., Yagi, M., Okizuka, Y. et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J Hum Genet 55, 379–388 (2010). https://doi.org/10.1038/jhg.2010.49
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.49
Keywords
This article is cited by
-
NGS-based targeted sequencing identified six novel variants in patients with Duchenne/Becker muscular dystrophy from southwestern China
BMC Medical Genomics (2023)
-
Benchmarking splice variant prediction algorithms using massively parallel splicing assays
Genome Biology (2023)
-
Cell-Based and Gene-Based Therapy Approaches in Neuro-orthopedic Disorders: a Literature Review
Regenerative Engineering and Translational Medicine (2023)
-
Genetic Analysis of Forty MLPA-Negative Duchenne Muscular Dystrophy Patients by Whole-Exome Sequencing
Journal of Molecular Neuroscience (2022)
-
Small mutations in Duchenne/Becker muscular dystrophy in 164 unrelated Polish patients
Journal of Applied Genetics (2021)
