
Abstract
Using exome sequencing we searched for the genetic cause of autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy (ARVC) in a Swedish family. A heterozygous C-to-T transition, c.1255C>T, p.Pro419Ser in the desmin gene on chromosome 2q35, was identified. Previous studies had demonstrated linkage to chromosome 10q22.3, but no causative mutation had been found in that region. Sanger sequencing of DNA from 17 family members confirmed the heterozygous c.1255C>T desmin mutation in seven out of ten family members that had been classified as affected in the previous study. Our new results demonstrate the usefulness of next-generation sequencing, and the diagnostic difficulties with some forms of dominantly inherited muscle diseases as they can display a wide clinical and morphological variability even within a given family.
Similar content being viewed by others

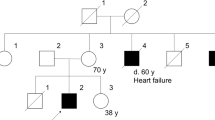
Introduction
Myofibrillar myopathies (MFMs) constitute a subgroup of muscular dystrophies defined by their similar morphological features beginning with disintegration of the Z-disc followed by accumulation of myofibrillar degradation products and degeneration of the muscle fibers ultimately leading to fibrosis. Clinically, MFMs are typically manifested by progressive proximal and/or distal muscle weakness and are frequently associated with cardiomyopathy, but the clinical picture is variable and depends on the mutated gene.1 MFMs to date have been associated with mutations in the Z-disc associated proteins (desmin, αB-crystallin, myotilin, Z-band alternatively spliced PDZ motif-containing protein (ZASP), filamin C and Bag3).1, 2
In this report the genetic cause of autosomal dominant myofibrillar myopathy (MFM) with arrhythmogenic right ventricular cardiomyopathy (ARVC) in a Swedish family was investigated. We identified the causative missense mutation in exon 7 of the desmin gene (DES) by performing exome sequencing in one family member and confirming the disease-associated mutation in other affected family members by Sanger sequencing.
Patients and methods
Patients
Seventeen individuals belonging to a Swedish family with MFM were available for investigation. Clinical and morphological features of affected family members have been reported in a previous study in which multipoint peak LOD score of 3.06 between markers D10S605 and D10S215 suggested linkage to chromosome 10q22.3.3 There was variable onset and severity of skeletal muscle and cardiac manifestations. The muscle weakness was axial in mildly affected, axial and distal in moderately affected or generalized in severely affected patients. None of the patients had ptosis, ophthalmoplegia, dysarthria or dysphagia. Muscle biopsy specimens showed myopathic changes, rimmed vacuoles, and accumulation of desmin, dystrophin and other proteins. Granulofilamentous changes and disorganization of myofibrils were observed on electron microscopic examination. Some biopsies showed only minor alteration. Three men had ARVC, two of them died at ages 59 and 63 years. There was severe fibrofatty infiltration of the right ventricle in one man at autopsy.3 Since the original study in 19993 one man with MFM and ARVC died at age 39. One woman developed atrial fibrillation at age 59 years.
Exome sequencing and bioinformatics
One family member with definite morphological evidence of MFM was selected for exome sequencing. Genomic DNA was isolated from a blood sample by standard methods. Five micrograms of genomic DNA was enriched using the Agilent SureSelect Human All Exon 38 Mb kit (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer's protocol and subsequently sequenced using Illumina HiSeq2000 following the manufacturer's instructions (Illumina, San Diego, CA, USA). Base calling was performed by the Illumina pipeline with default parameters. All the raw reads were aligned to the reference human genome (UCSChg19) using the Burrows–Wheeler alignment and SNPs and indels were identified using the Genome Analysis Tool Kit.4, 5 Variant filtration was made by focusing on SNPs and small indels in a region on 10q22.3 between the markers D10S605 (chr10:79.25 Mb) and D10S215 (chr10:89.47 Mb) and an additional 24 proteins involved in the Z-disc and the intermediate filament (Supplementary Table S1).2
Genetic analysis
Sanger sequencing was performed using the standard techniques of PCR amplicons with primers 5′-CTGGGATTACAGCTGGGG-3′ and 5′-CAGGGTCCCATCCTCCC-3′ from genomic DNA to confirm the presence and identity of a variant in the DES (NM_001927.3) after it had been identified by exome sequencing.
Results
Exome sequencing and bioinformatics
We identified in the whole exome 25666 SNPs and indels of them were 23694 reported in dbSNP131. The SNP and indel searches in the region on chromosome 10q flanked by the microsatellite markers D10S605 and D10S21 identified 52 SNPs and no indels. We excluded 50 known variants reported in dbSNP version 131 and two novel SNPs because they were synonymous. In Z-disc and intermediate filament associated proteins (Supplementary Table S1) we identified 19 SNPs and one indel. We excluded 18 of the SNPs and the indel reported in dbSNP version 131. The remaining novel non-synonymous SNP was then the probable causative mutation, a heterozygous mutation, c.1255C>T, p.Pro419Ser, in exon 7 in the DES on chromosome 2 (NM_001927.3) (Table 1).
Genetic analysis
Sanger sequencing of 17 family members demonstrated the heterozygous DES mutation c.1255C>T, p.Pro419Ser in exon 7 on chromosome 2 in 7 of the family members.
Discussion
By exome sequencing we have identified a mutation in the DES on chromosome 2 to be responsible for MFM in a large Swedish family. This was the first family reported with MFM in association with ARVC.3 Because of its putative unique map position on chromosome 10q22.3 this disease has been named ARVC7 (OMIM entry 609160). The ZASP gene located in the vicinity of the 10q22.3 region has been reported to be mutated in patients with MFM and some of them had cardiac involvement.6 However, mutation screening of ZASP and other genes in the 10q22.3 region was negative in our family.7
Using an unbiased approach, next-generation sequencing technology on a genome-wide scale we succeeded in finding a heterozygous C-to-T transition, c.1255C>T, p.Pro419Ser, in exon 7 of DES. Sanger sequencing of the corresponding DES region in 17 family members confirmed the heterozygous c.1255C>T mutation in seven out of ten family members that had been classified as affected in the previous study. This Pro-to-Ser mutation at codon position 419 in the tail region of desmin has already been discovered in unrelated patients suffering from MFM supporting the pathogenicity of the mutation.8 The DES was sequenced in 2004 in an affected member of this family, but the mutation was not detected (data unpublished).
Five family members classified as affected in the previous study were asymptomatic but showed trunk-muscle weakness and myopathic electromyography changes.3 The muscle biopsy alterations in three out of these five turned out to be unspecific and not sufficient for a diagnosis of MFM. We have not diagnosed a specific muscular disease in these three asymptomatic individuals without desmin storage. Only patients detected with desmin storage carried the mutation, including two patients who were asymptomatic with weakness of the trunk.
Desmin myopathy is also known to be associated with cardiomyopathy.9 Right ventricular cardiomyopathy and ARVC that were present in individuals of this family have been linked to DES mutations in recent years.10, 11, 12, 13
In conclusion, we have, by means of exome sequencing, identified the causative DES mutation in a family with MFM and ARVC. Our results demonstrate diagnostic difficulties with some forms of dominantly inherited muscle diseases as they can display a wide clinical and morphological variability even within a given family. We now correct the locus of ARVC7 to DES on chromosome 2q35.
References
Selcen D : Myofibrillar myopathies. Neuromuscul Disord 2011; 21: 161–171.
Laing NG, Nowak KJ : When contractile proteins go bad: the sarcomere and skeletal muscle disease. Bioessays 2005; 27: 809–822.
Melberg A, Oldfors A, Blomstrom-Lundqvist C et al: Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy linked to chromosome 10q. Ann Neurol 1999; 46: 684–692.
Li H, Durbin R : Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
DePristo MA, Banks E, Poplin R et al: A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43: 491–498.
Selcen D, Engel AG : Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann Neurol 2005; 57: 269–276.
Kuhl A, Melberg A, Meinl E et al: Myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy 7: corroboration and narrowing of the critical region on 10q22.3. Eur J Hum Genet 2008; 16: 367–373.
Olive M, Armstrong J, Miralles F et al: Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromuscul Disord 2007; 17: 443–450.
Goldfarb LG, Dalakas MC : Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest 2009; 119: 1806–1813.
Maass K : Arrhythmogenic right ventricular cardiomyopathy and Desmin: another gene fits the shoe. Heart Rhythm 2010; 7: 1065–1066.
Otten E, Asimaki A, Maass A et al: Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm 2010; 7: 1058–1064.
van Tintelen JP, Van Gelder IC, Asimaki A et al: Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm 2009; 6: 1574–1583.
Klauke B, Kossmann S, Gaertner A et al: De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet 2010; 19: 4595–4607.
Acknowledgements
The study was supported by grants from the Swedish Research Council project no. 07122 and the Selander Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Hedberg, C., Melberg, A., Kuhl, A. et al. Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy 7 is caused by a DES mutation. Eur J Hum Genet 20, 984–985 (2012). https://doi.org/10.1038/ejhg.2012.39
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2012.39
Keywords
This article is cited by
-
Genotype–phenotype Correlates in Arrhythmogenic Cardiomyopathies
Current Cardiology Reports (2022)
-
Arrhythmogenic Cardiomyopathy: Mechanisms, Genetics, and Their Clinical Implications
Current Cardiovascular Risk Reports (2021)
-
Molecular insights into cardiomyopathies associated with desmin (DES) mutations
Biophysical Reviews (2018)
-
Limb-girdle muscular dystrophies — international collaborations for translational research
Nature Reviews Neurology (2016)
-
Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia (ARVC/D): Review of 16 Pediatric Cases and a Proposal of Modified Pediatric Criteria
Pediatric Cardiology (2016)
