
Abstract
Rett syndrome is a severe neurodegenerative disorder characterized by acquired microcephaly, communication dysfunction, psychomotor regression, seizures and stereotypical hand movements. Mutations in methyl CpG binding protein 2 (MECP2) are identified in most patients with classic Rett syndrome. Genetic studies in patients with a Rett variant have expanded the spectrum of underlying genetic etiologies. Recently, a deletion encompassing several genes in the long arm of chromosome 14 has been associated with the congenital Rett-syndrome phenotype. Using array-based comparative genomic hybridization, we identified a 3-year-old female with a Rett-like syndrome carrying a de novo single-gene deletion of FOXG1. Her presentation included intellectual disability, epilepsy and a Rett-like phenotype. The variant features included microcephaly at birth and prominent synophrys. Our results confirm that congenital Rett syndrome can be caused by copy-number variation in FOXG1 and expand the clinical phenotypic spectrum of FOXG1 defect in humans.
Similar content being viewed by others


Introduction
Rett syndrome is a severe neurodegenerative disorder first described by Andreas Rett in 1966.1 It is classically described by acquired microcephaly (between 3 months and 4 years of age), psychomotor regression, mental retardation, seizures and stereotypic hand movements.2 Variation of clinical severity in Rett-syndrome phenotype has emerged with milder or more severe phenotypes being identified.2, 3, 4, 5, 6, 7 In 2002, the diagnostic criteria for the variant Rett syndrome were revised6 to help achieve a better diagnostic categorization between classic Rett syndrome and variant Rett syndrome. Five clinical variants have been distinguished: (i) the infantile-seizure-onset variant, (ii) the congenital variant, (iii) the ‘forme fruste’, (iv) the late-regression variant and (v) the preserved-speech variant.5 Molecular gene testing in females with the classic Rett-syndrome phenotype identifies a mutation in the methyl CpG binding protein 2 (MECP2) gene in 80% of cases.8, 9 Unlike the classic Rett syndrome, the genetic etiologies of variant Rett syndrome are heterogeneous and in recent years numerous reports of new mutations have emerged. Mutations in the MECP2 and CDKL5 genes have been identified (RettBASE, http://mecp2.chw.edu.au, 2009). The latter in individuals with a Rett-syndrome-like phenotype and early-onset seizures.10
The congenital variant is characterized by hypotonia and developmental delay beginning earlier than in the classic Rett syndrome.11, 12 The majority of children with a congenital variant of Rett syndrome do not harbor MECP2 or CDKL5 mutations.10, 13 Recent advances in genomic screening have linked this variant to the forkhead box G1 gene (FOXG1) that encodes forkhead box protein G1.3 Here, we describe a young girl presenting with a variant Rett-syndrome phenotype associated with a de novo selective deletion of the FOXG1 gene previously not described.
Case report
The proband is a 3-year-old female, born to non-consanguineous parents (Figure 1a and b). There is no history of teratogen exposure. The mother has ulcerative colitis, which was treated with sulfasalazine daily throughout the entire pregnancy. Antenatal ultrasound at 19 weeks showed microcephaly, however, a subsequent ultrasound showed the head circumference to be within normal limits. Delivery was at two days post term, through caesarean section for failure of progression of labour. The infant did not require resuscitation. APGAR scores were 9 and 9 at 1 and 5 min, respectively. Birth weight was 3940 g (75th centile), length was 50 cm (50th centile) and her head circumference 32.5 cm (3–5th centile) (Figure 1c). The neonatal course was complicated by hyperbillirubinemia treated with phototherapy and feeding difficulties. Developmental delay was noticed at 4 months of age. At sixteen months, she could not sit without support, she could not roll or crawl and was non-verbal. At 11 months, she had developed seizures consisting of partial complex and myoclonic seizures. She is currently treated with phenobarbital after failed anti-convulsants, including valproic acid, clobazam and carbamazepine. At present she sits with assistance, can roll and uses a raking grasp. She is non-verbal and will occasionally make eye contact. She has bilateral cortico–visual impairment, with the left eye having esotropia and the right eye homonymous hemianopsia. She has stereotypic hand movements (batting and wringing) and grinds her teeth. The patient presents daytime and night-time episodes of inexplicable screaming that switch to laughing without obvious reason. She is affected by chronic constipation and gastroeosophageal reflux. A gastrostomy was placed for nutritional supplementation. There is no history of regression or loss of social interaction. The family history is unremarkable, including no known neonatal deaths, chromosomal abnormalities, seizures, epilepsy or spontaneous abortions.

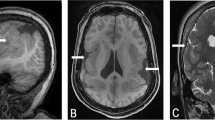
Loss of one copy of FOXG1 leads to a Rett-variant phenotype. (a) Picture of the patient taken at 3 years of age showing the synophrys and a pointed chin. Note the posturing of the hands. (b) Lateral view of the patient. Note the brachycephaly. (c) Occipito–frontal head-circumference measurements show onset of microcephaly before 1 year of age. (d) Sagittal T1-weighted MRI carried out at 6 months of age illustrates the brachycephaly. (e) Axial flair T2-weighted MRI carried out at 6 months of age shows normal myelination for age and absence of cerebral malformation.
On examination at three years of age, her head circumference was 41.5 cm (≪2nd centile) (Figure 1c), her weight was 13 kg (25th centile) and her height was 95 cm (50th tile). The following dysmorphic features were observed: mild synophrys, low-set ears, pointed chin and bulbous upturned nose. Cranial nerve examination showed reactive pupils, normal fundi and roving eye movements with no evidence of tracking. The palate was high arched and the filtrum flat. There was hypotonia of the neck and trunk. Dyskinetic choreoathetoid movements of the face, trunk and limbs were noted. She was hyper-reflexic, bilaterally and symmetrically. She showed flexor plantar responses bilaterally and there was no clonus. No scoliosis or lower-limb atrophy was noted.
Magnetic resonance imaging (MRI) of the head was carried out at 6 months of age and showed microcephaly with appropriate structure and myelination. (Figure 1d and e) An electroencephalogram (EEG) was carried out at 16 months of age and showed a slow, theta frequency, background with epileptiform discharges from the left temporal lobe. Several other EEGs were carried out and showed diffusely slow, theta frequency, backgrounds. Brainstem auditory-evoked responses were normal. A metabolic screen, including quantitative serum amino acids, urine organic-acid profile, serum lactate, carnitine and creatine-kinase levels were all normal.
Chromosome and array-based comparative genome hybridization analysis
Informed consent was obtained from the patient's parents. Karyotyping was carried out according to routine protocols. Twenty metaphases with 550-band resolution were analyzed. An array-based comparative genomic hybridization analysis using 105 K CMA OLIGO V7.2, was carried out at the Baylor College of Medicine (http://www.bcm.edu/geneticlabs/cma/tables.html) by the Kleberg Cytogenetics Laboratory (Houston, TX, USA, www.bcmgeneticlabs.org). Confirmatory FISH analyses were carried out using the BAC clones RP11-30H9 and RP11-445P17 also at the Baylor College of Medicine. The karyotype was found to be 46XX. CMA V7.2 OLIGO showed a minimum of 2.529 Mb to a maximum of 2.594 Mb loss of copy number in the 14q12 region. (Figure 2) The proximal breakpoint was mapped between 26 323 511 and 26 355 719 and the distal breakpoint between 28 884 679 and 28 917 989. The genes localized to this region include FOXG1 and C23orf14. (Figure 2) In addition, a minimum of 0.246 Mb to a maximum of 0.318 Mb loss in the 10.15 region of the short arm of chromosome 10 was identified. The proximal breakpoint was mapped between 5 080 373 and 5 127 042 and the distal breakpoint between 5 373 173 and 5 398 534. The genes localized to that location include AKR1C3, AKR1CL1 and AKR1C4. FISH analysis using the BAC clones RP11-30H9 and RP11-445P17 confirmed the deletion. FISH analysis of parental chromosomes showed a paternal loss in the 10.15 region of the short arm of chromosome 10 similar to the proband's. No evidence for a lesion on chromosome 14 was detected in either parent.

Cartoon representation of chromosome 14, region 26 000 000–29 500 000. **represents the 2.594-Mb deletion identified in the case report. This region includes both the FOXG1 gene and C14orf23 gene. The genes PRKD1 and NOVA as shown, are not affected by the deletion. The location of the probe RP11-30H9 is also identified. Figure adapted from www.ensemble.org (chromosome 14: 26 000 000–29 500 000).
Discussion
Given the proband's clinical history of developmental delay, hand stereotypies, postnatal deceleration of head growth in the context of a normal prenatal and perinatal history we first hypothesized a Rett spectrum disorder. On the basis of her congenital microcephaly, her early psychomotor delay and early-onset epilepsy she did not meet the criteria for the classical Rett-syndrome phenotype. However, she does fulfill the diagnostic criteria for the variant Rett syndrome.6 Main diagnostic criteria include reduction of hand skills, repetitive hand stereotypies and deceleration of head growth from early childhood. Supportive criteria include air swallowing, bruxism, abnormal locomotion, inexplicable episodes of screaming and laughing, as well as night-time screaming. The early onset of her symptoms and microcephaly at birth classifies her as the congenital variant. Comparative genomic hybridization identified a loss of 0.246–0.318 Mb at chromosome 10p15.1 as well as a 2.529–2.594 Mb loss on chromosome 14q12. The loss of genetic material on the short arm of chromosome 10 includes the AKR1C3, AKR1CL1 and AKR1C4 genes. These genes are members of the aldo–keto reductase family 1 and have not been reported to be associated with developmental delay, microcephaly or a Rett-like phenotype. Importantly, parental FISH analysis showed a similar chromosome 10 deletion in the proband's father. The father is entirely asymptomatic showing no evidence of dysmorphic features or neurological impairment. Therefore, we believe the loss of material on chromosome 10 is not contributing to the patient's clinical phenotype. Conversely, the loss on chromosome 14 was not identified in the parents and was therefore considered to be responsible for the patient's symptoms.
Lesions along the long arm of chromosome 14 have been linked to developmental delay and epilepsy. Indeed, Kamnasaran et al14 reported nine cases of large deletions or translocations, involving the 14q11–q21 region. The clinical spectrum included global developmental delay, corpus callosal abnormalities, hypotonia, delayed myelination, seizures and microcephaly. The unbiased exploration of genetic lesions leading to Rett syndrome-like phenotype has emerged with the use of array-based comparative genomic hybridization (aCGH). Recently two cases of congenital variant of Rett syndrome were identified to have a deletion of long arm of chromosome 14, which included forkhead box G1 (FOXG1). Table 1 shows a comparative summary of clinical features linked to FOXG1 deletion or mutation. Bisgaard et al15 reported an 11-month-old female with psychomotor retardation, seizures, microcephaly and dysmorphic features. In addition to FOXG1, the deleted region contains PRKD1. Papa et al16 reported a 7-year-old girl with severe mental retardation, epilepsy, microcephaly and Rett-like features. The deleted region contains FOXG1, PRKD1, SCFD1, COCH and STRN3 genes. The link between Rett variant and FOXG1 is supported by previous reports of patients without complete deletion of FOXG1 but with lesions in its coding region. Shoichet et al17 reported the case of a female patient showing a severe cognitive disability associated with complete agenesis of the corpus callosum, acquired microcephaly, seizures and tetraplegia. The patient carried a balanced de novo translocation t(2;14)(p22;q12), together with a neighbouring 720-kb inversion in chromosome 14q12 that included FOXG1. More recently, Ariani et al3 identified two girls with congenital Rett variant with mutation in FOXG1. The first case was a 22-year-old woman with a congenital variant of Rett syndrome and a point mutation in FOXG1. This point mutation introduced a stop codon that is predicted to disrupt DNA binding by disrupting the forkhead domain. The second patient was a 7-year-old girl with a 1-bp deletion in the FOXG1 gene leading to the loss of the JARID1B-interacting domain and misfolding of the region responsible for groucho binding. Both patients had intellectual disability associated with corpus callosum hypoplasia, microcephaly, stereotypic activities, occasional abnormal breathing patterns and bruxism.
The FOXG1 gene has been shown to have a vital role in the formation of the developing brain. It encodes a transcriptional repressor protein with expression restricted to fetal and adult brain, and testis.18 In the developing brain, FOXG1 expression is restricted to the telencephalic neuroepithelium and the nasal half of the retina and optic stalk. FOXG1 defines the region that will become the telencephalon.19 Moreover, following FOXG1 expression the single-cell thick telencephalon primordium expands into a multicellular, subdivided tissue. This is accomplished in collaboration with sonic hedgehog through fibroblast growth factor (FGF) production. Hanashima20 showed that the Cajal–Retzius cells, the earliest neurons established in cortical layering, are suppressed by FOXG1. Conversely, FOXG1-null mice have been observed to have an excess of Cajal–Retzius neuron production in the cortex. Loss of function of the orthologous mouse gene FOXG1 leads to a severe decrease in the cerebral-hemisphere size and early depletion of progenitor cells.21 Owing to its role in the developing brain it is not surprising that the disruption of the FOXG1 gene leads to the clinical phenotype of developmental delay. Interestingly, Ariani et al3 found overlapping expression of FOXG1 and MECP2 in differentiating forebrain cortex. In addition, they have also shown colocalization in NIH3T3 cells and primary neurons. Although their exact interaction still remains to be defined, this may suggest a common pathway or interaction at crucial points during brain development.
To our knowledge this case represents the third case reported of a patient with a deletion smaller than 3.5 Mb involving the band 14q12 but the first involving selectively FOXG1 leading to congenital Rett variant. In addition to reproducing the previous findings of severe developmental delay, epilepsy and generalized dyskinetic movements, this report expands the clinical spectrum of FOXG1 mutations in humans. New clinical features observed in our case are early psychomotor retardation, the facial feature of mild synophrys, absence of callosal agenesis or hypoplasia and suspected microcephaly in utero. In light of this, we would suggest that FOXG1 sequencing in suspected cases of congenital and other variant Rett syndrome patients.
References
Rett A : [On a unusual brain atrophy syndrome in hyperammonemia in childhood]. 1966; 166: 723–726.
Hagberg B : Rett syndrome: clinical peculiarities and biological mysteries. Acta Paediatr 1995; 84: 971–976.
Ariani F, Hayek G, Rondinella D et al: FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet 2008; 83: 89–93.
Hagberg B : Clinical delineation of Rett syndrome variants. Neuropediatrics 1995; 26: 62.
Hagberg BA, Skjeldal OH : Rett variants: a suggested model for inclusion criteria. Pediatr Neuroly 1994; 11: 5–11.
Hagberg B, Hanefeld F, Percy A, Skjeldal O : An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol 2002; 6: 293–297.
Percy AK : Rett syndrome. Current status and new vistas. Neurol Clin 2002; 20: 1125–1141.
Dragich J, Houwink-Manville I, Schanen C : Rett syndrome: a surprising result of mutation in MECP2. Hum Mol Genet 2000; 9: 2365–2375.
Shahbazian MD, Zoghbi HY : Molecular genetics of Rett syndrome and clinical spectrum of MECP2 mutations. Curr Opin Neurol 2001; 14: 171–176.
Scala E, Longo I, Ottimo F et al: MECP2 deletions and genotype-phenotype correlation in Rett syndrome. Am J Med Genet 2007; 143A: 2775–2784.
Rolando S : Rett syndrome: report of eight cases. Brain Dev 1985; 7: 290–296.
Shimamura K, Martinez S, Puelles L, Rubenstein JL : Patterns of gene expression in the neural plate and neural tube subdivide the embryonic forebrain into transverse and longitudinal domains. Dev Neurosci 1997; 19: 88–96.
Erlandson A, Samuelsson L, Hagberg B, Kyllerman M, Vujic M, Wahlstrom J : Multiplex ligation-dependent probe amplification (MLPA) detects large deletions in the MECP2 gene of Swedish Rett syndrome patients. Genet Test 2003; 7: 329–332.
Kamnasaran D, O'Brien PC, Schuffenhauer S et al: Defining the breakpoints of proximal chromosome 14q rearrangements in nine patients using flow-sorted chromosomes. Am J Med Genet 2001; 102: 173–182.
Bisgaard AM, Kirchhoff M, Tumer Z et al: Additional chromosomal abnormalities in patients with a previously detected abnormal karyotype, mental retardation, and dysmorphic features. Am J Med Genet 2006; 140: 2180–2187.
Papa FT, Mencarelli MA, Caselli R et al: A 3 Mb deletion in 14q12 causes severe mental retardation, mild facial dysmorphisms and Rett-like features. Am J Med Genet 2008; 146A: 1994–1998.
Shoichet SA, Kunde SA, Viertel P et al: Haploinsufficiency of novel FOXG1B variants in a patient with severe mental retardation, brain malformations and microcephaly. Hum Genet 2005; 117: 536–544.
Murphy DB, Wiese S, Burfeind P et al: Human brain factor 1, a new member of the fork head gene family. Genomics 1994; 21: 551–557.
Tao W, Lai E : Telencephalon-restricted expression of BF-1, a new member of the HNF-3/fork head gene family, in the developing rat brain. Neuron 1992; 8: 957–966.
Hanashima C, Li SC, Shen L, Lai E, Fishell G : Foxg1 suppresses early cortical cell fate. Science 2004; 303: 56–59.
Xuan S, Baptista CA, Balas G, Tao W, Soares VC, Lai E : Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron 1995; 14: 1141–1152.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jacob, F., Ramaswamy, V., Andersen, J. et al. Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature. Eur J Hum Genet 17, 1577–1581 (2009). https://doi.org/10.1038/ejhg.2009.95
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.95
Keywords
This article is cited by
-
FOXG1 Regulates PRKAR2B Transcriptionally and Posttranscriptionally via miR200 in the Adult Hippocampus
Molecular Neurobiology (2019)
-
Partial monosomy14q involving FOXG1 and NOVA1 in an infant with microcephaly, seizures and severe developmental delay
Molecular Cytogenetics (2016)
-
Dysregulation of FOXG1 by ring chromosome 14
Molecular Cytogenetics (2015)
-
Platelet defects in congenital variant of Rett syndrome patients with FOXG1 mutations or reduced expression due to a position effect at 14q12
European Journal of Human Genetics (2013)
-
14q12 microdeletions excluding FOXG1 give rise to a congenital variant Rett syndrome-like phenotype
European Journal of Human Genetics (2013)
