
Abstract
We report on three patients with split hand/foot malformation type 1 (SHFM1). We detected a deletion in two patients and an inversion in the third, all involving chromosome 7q21q22. We performed conventional chromosomal analysis, array comparative genomic hybridization and fluorescence in situ hybridization. Both deletions included the known genes associated with SHFM1 (DLX5, DLX6 and DSS1), whereas in the third patient one of the inversion break points was located just centromeric to these genes. These observations confirm that haploinsufficiency due to either a simultaneous deletion of these genes or combined downregulation of gene expression due to a disruption in the region between these genes and a control element could be the cause of the syndrome. We review previously reported studies that support this hypothetical mechanism.
Similar content being viewed by others

Introduction
Split hand/foot malformation (SHFM; OMIM 183600) is a congenital limb defect characterized by a deep median cleft of the hand/foot due to absence or hypoplasia of the central rays.1 The overall prevalence of SHFM is approximately 1:18 000 newborns.2 It can occur either as part of a syndrome or as an isolated malformation.2
Five types of non-syndromic SHFM that have been mapped to different loci can be distinguished genetically.2, 3 SHFM2 maps to Xq26 and SHFM5 to 2q24q31. Although the disease genes for SHFM2 and SHFM5 have not been identified yet, a function has been suggested for the HOXD genes in SHFM5.2 SHFM3 is associated with genomic alterations on chromosome 10q24q25, which has been shown to result in complex gene rearrangements in the DACTYLIN gene.4 SHFM4 (3q27) results from mutations in the p63 gene.5, 6 The most common form, SHFM1, is caused by chromosomal rearrangements involving 7q21q22.7, 8 SHFM1 has been reported to be an autosomal dominant trait with reduced penetrance and variable expression.9 It is associated with deafness in 35% of patients10 (OMIM 220600), which can be accompanied by a Mondini malformation of the inner ear. A microdeletion of 7q21q22 has refined the critical SHFM1 region to 0.9 Mb.11 This interval includes the candidate genes DSS1, and the distalless-related homeogenes DLX5 and DLX6, which are members of the Wnt pathway that is important in limb development. Animal models in which Dlx5 and Dlx6 have been targeted for deletion exhibit an SHFM phenotype only if both the Dlx5 and Dlx6 genes are deleted. In patients with SHFM, no mutations have been found so far in these genes.1, 2
We report here on three patients with SHFM1 who have aberrations of chromosome 7q21q22; two patients have a deletion involving DLX5 and DLX6, and one patient has an inversion break point in this region. We explore the underlying mechanism of the pathogenesis of SHFM1 in these patients and review previously reported studies that would support this mechanism.
Methods
Patients
Three patients with a combination of hand defects and chromosome 7q aberration were clinically studied. Patient 3 has been reported before at the age of 2 years.9 The patient revisited our clinic at age 17 years in 2008.
Conventional chromosomal analysis
Peripheral blood lymphocytes were obtained from the patients and their parents. Whole blood lymphocytes were cultured with phytohemagglutinin and cells were harvested using standard cytogenetic techniques. Chromosomes were G-banded using pancreatin and Giemsa.
Array comparative genomic hybridization analysis
The aberrations were characterized by array comparative genomic hybridization (array-CGH) using either a 244K oligo array or a 105K oligo array (design IDs 014693 and 019015, respectively; Agilent Technologies Inc., Santa Clara, CA, USA). Procedures were carried out according to the manufacturer's protocols and the data were processed using Feature Extraction V.9.1 and CGH analytics V.3.4.27 as provided by Agilent Technologies Inc. Only deletions larger than 150 kb and duplications sized more than 200 kb were called. The Toronto database of Genomic Variants (http://projects.tcag.ca/variation/) was used to exclude well-known benign copy number variants.
Fluorescence in situ hybridization analysis
Fluorescence in situ hybridization (FISH) analysis was used to precisely define the 7q inversion break point of patient 3. Bacterial artificial chromosome (BAC) clones were selected from the human library RPCI-11, according to the UCSC Human Genome Assembly (freeze March 2006) and provided by the Wellcome Trust Sanger Institute (http://www.sanger.ac.uk). The following BAC clones were used: P164D18 (7pter); RP11-51L23, RP11-70K3, RP11-71F18 (7p21.1); RP11-837N16, RP11-77D17, RP11-384P11, RP11-266G22, RP11-159D2 (7q21.3) and RP11-93F2 (7qter).
BAC DNA was labelled with biotine- and digoxigenin-11-dUTP using Nick translation. Slides were hybridized at 37°C overnight and fluorescently labelled with fluorescein isothiocyanate and Texas Red, respectively.
Results
Patient 1
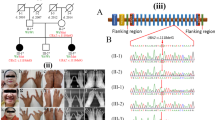
The first patient was a girl, born with an intra-uterine growth retardation (weight −3 SD, length −3 SD, head circumference −2 SD). She was the third child of healthy, unrelated parents. Her brother and sister were both healthy. Congenital anomalies of the hands and feet were noted immediately after birth. Physical examination showed typical central reduction defects of all four extremities. The second and third rays were missing on both hands, and there was syndactyly of the fourth and fifth fingers. Only one ray was present in both feet (Figure 1a). Because of the abnormal position of the feet and a non-functional ankle joint, the feet were amputated and prostheses made. The age at last examination was 3.5 years (weight −2 SD, length −5 SD (corrected for prostheses), head circumference −2.5 SD). She had some minor facial dysmorphisms (frontal bossing, micrognathia, small dysplastic ears and a long philtrum). She has congenital deafness due to aplasia of the cochlear nerves, which has been detected by MRI.

Hands and feet of (a) patient 1 showing typical central reduction defects of all four extremities, with only one ray present in both feet; (b) patient 2 showing hypoplastic and hyperconvex nails, and on the index finger only the ulnar part of the nail, on the right foot a cutaneous syndactyly of the second and third toes until the proximal interphalangeal joints with an abnormal position of the second toe and a broad hallux; (c) patient 3 showing central reduction defects of all four extremities, and an arteriovenous malformation on the right hand.
Chromosomal investigation showed a de novo deletion of the long arm of chromosome 7: 46,XX,del(7) (q21.13q22.1). The deletion was further characterized by the 105K oligo array-CGH and had a maximal size of 8.66 Mb, located at 89.87–98.52 Mb from the p-telomere of chromosome 7 (Figure 2). No other clinically relevant copy number variants were detected.

Overview of chromosome 7q with the deletions of patients 1 and 2 and the inversion break point of patient 3. The region of interest has been magnified and again shows the deletions of patients 1 and 2, and the inversion break point in 7q21.3 of patient 3. Also shown are the locations of DLX5, DLX6, DSS1, DYNC1I1, SLC25A13, CUTL1 and FZD1.
Patient 2
The second patient was a 2-month-old girl, born after an uneventful pregnancy and delivery (weight −1.5 SD, length +0.5 SD, head circumference 0 SD). She was the first child of healthy, unrelated parents. The family history did not include any congenital malformations. On day 7 she was admitted to the paediatrics clinic because of severe feeding difficulties, insufficient sucking reflex, tracheomalacia and hypotonia. Physical examination showed some facial dysmorphisms, including low set ears with a folded upper part of the helices and an additional groove, upslanting and short palpebral fissures, small mouth and retrognathia. She had hypoplastic and hyperconvex nails on both hands. Only the ulnar part of the nails was developed on the index fingers. The right foot showed cutaneous syndactyly of the second and third toes until the proximal interphalangeal joints and an abnormal position of the second toe. Both halluces were broad and their nails displayed longitudinal ridges. Both forefeet were slightly inverted (Figure 1b). X-ray studies of the hands and feet showed normal morphology in the hands, but a long and slender proximal phalanx of the second toe of the right foot and broad phalanges of the first toes, with a possible abortive duplication of the terminal phalanx of the right hallux. Audiological examination revealed severe bilateral sensorineural hearing loss. Cardiac and abdominal ultrasound did not reveal any structural heart or renal defects.
Chromosomal investigation showed a de novo deletion of chromosome 46,XX,del(7)(q21.11q21.3). The deletion was further characterized by the 105K oligo array-CGH and had a maximal size of 12.94 Mb, located at 83.88–96.81 Mb from p-telomere on chromosome 7 (Figure 2). No other clinically relevant copy number variants were detected.
Patient 3
This boy was born after an uneventful pregnancy as the first child of healthy, unrelated parents. The family history was unremarkable. Physical examination at the age of 2 days showed typical central reduction defects of all four limbs. The third finger on the left hand and the second, third and fourth fingers on the right hand were missing. The fifth ray of the right hand was swollen and red-blue in colour so arteriographic studies of the right arm were performed and revealed an arteriovenous malformation on the ulnar ray of the hand. Both feet showed absence of the second, third and fourth toes. The right foot also had a deformity of the first toe, which was in a retroflexed position. X-ray studies confirmed the reduction defects and showed a bifid distal thumb phalanx of the right hand. At the age of 2 years the boy showed a normal cognitive development. The arteriovenous malformation was partially involuted.9
The boy was re-evaluated at age 17 years (Figure 1c) when no dysmorphic features were evident. He used his hands and feet well and was able to write with his left hand. He had undergone two surgical corrections on his right foot to correct a hyperextensed position. At age 7 years he had been diagnosed with PDD-NOS. He attended normal secondary school, although at a lower level than would be expected given his parents' educational level. There was no deafness, although his hearing has not been investigated with an audiological examination.
Chromosomal investigation showed a de novo pericentric inversion of chromosome 7: 46,XY,inv(7)(p22q21.3). A normal male pattern was found by high-resolution 244K oligo array-CGH, indicating no imbalance genome wide and particularly not at the break-point regions 7p22 and 7q21.3. FISH was performed to define the break point in 7q more precisely and the probe RP11-837N16 appeared to span this break point. This probe maps to the region 95.53–95.72 Mb, locating the break point in 7q21.3, situated just beside the DLX5, DLX6 and DSS1 genes (Figure 2).
Review of literature and Discussion
SHFM1 is associated with deletions of variable extent on chromosome 7q21q22, minimally including DSS1 and the distalless-related homeogenes DLX5 and DLX6. Chromosome 7q alterations resulting in SHFM reported thus far are summarized in Figure 3.1, 23, 32

Overview of literature regarding split/hand foot due to chromosome 7 aberrations.1, 23, 32 From top to bottom: distance in Mb from p-telomere; microsatellite markers used to map previous patient break points; smallest region of overlap (SRO) defined by the distal deletion break point in patient D5 (Crackower et al14) and by the proximal deletion break point in patient D7 (Wieland et al11); FISH probes used to map patient 3 and patient B (Bernardini et al1) approximate translocation break points within the SRO; the location of the genes DSS1, DLX5, DLX6 and DYNC1I1.
Two of our patients (1 and 2) had deletions of chromosome 7q21q22, including the genes DLX5, DLX6 and DSS1 known to have a function in SHFM1. In mice, Dlx transcription factors have been shown to have a key function in the development and morphogenesis of the limb skeleton.3, 12 Expression of Dlx5 and Dlx6 has been detected in the apical ectodermal ridge of the embryonic limb buds. Double knockout of Dlx5 and Dlx6 in the mouse leads to ectrodactyly. Knockout mice that solely lack the Dlx5 gene or with double heterozygous knockout of both Dlx5 and Dlx6 do not display limb abnormalities. This suggests that the Dlx5 and Dlx6 genes are functionally redundant in controlling limb development in mice. In contrast to the situation in mice, concomitant haploinsufficiency of DLX5 and DLX6 seems to be the causative mechanism in humans. This difference in dosage sensitivity of DLX5 and DLX6 suggests that humans are much more sensitive to disruptions in these genes than the mouse heterozygotes.3, 10, 12 This same difference between mice and humans has been shown for LMX1B, for which haploinsufficiency in humans causes nail patella syndrome (OMIM 161200), whereas only complete knockout mice exhibit such a phenotype.13
The function of the DSS1 gene in limb formation, however, is less clear. It was named deleted in split-hand/split-foot 1 region (DSS1) because it was identified within the critical chromosomal region of SHFM1.14 The Dss1 gene encodes a highly conserved acidic protein, which is expressed in the branchial arches, genital tubercle and the developing limb buds in mice.14 In yeast, Sem1, the homologue of human DSS1, is involved in a cellular differentiation process.15 This suggests DSS1 could also be involved in the pathogenesis of SHFM. However, studies in mice with double knockout of Dlx5 and Dlx6 showed typical ectrodactyly,3, 12 whereas Dss1 expression was normal in these mutant mice.12, 16 This suggests downregulation of DLX5 and DLX6 is sufficient to cause SHFM whereas DSS1 expression is normal.
Our third patient had an inversion with one of the break points on chromosome 7q21.3 just next to the DLX5, DLX6 and DSS1 genes. In theory, the phenotype could have two explanations in this patient. First, there could be a new gene associated with SHFM disrupted by the inversion break points. The 7p region has never been associated with SHFM and was not further explored. The only known genes within the 7q break-point region are SLC25A13, a member of the solute carrier family 25, which has a function in the urea cycle,17 and DYNC1I1, cytoplasmic dynein intermediate chain 1 gene, which is a large microtubule-based motor protein not expressed in developing limb buds.18 These genes are therefore unlikely to explain our patient's phenotype. The more likely, second, option is that the inversion break point influences the expression of DLX5 and DLX6. It has been suggested that chromosomal rearrangements can separate critical regulatory elements from their corresponding genes. This mechanism is very likely to occur in SHFM1.7, 8 Thus, the phenotype of patient 3 might be explained by the physical separation of a control element from the DLX5 and DLX6 genes, causing reduced expression of both genes and resulting in the SHFM1 phenotype. The control element is probably located proximal to the 7q21.3 inversion break point.
The hypothesis of ‘functional haploinsufficiency’ is in line with two important observations. First, only the combined deletion of the Dlx5 and Dlx6 genes results in SHFM1.2, 3, 16 Second, the phenotype in SHFM4, due to p63 mutations, has been shown to be mediated through both Dlx5 and Dlx6.3 In p63−/− newborn mice, the hind limbs are absent, whereas the fore limbs are severely truncated in the distal segment. In these mice the expression of the Dlx5 and Dlx6 genes was significantly reduced. Lo Iacono et al3 provided evidence that p63 is genetically upstream of Dlx genes in the apical ectodermal ridge and thus alteration of the p63–Dlx pathway is the most likely molecular basis of both SHFM4 and SHFM1.
Okita et al19 showed presumptive evidence for an imprinting domain in the region of chromosome 7q21q31, with DLX5 only expressed from the maternal allele in brain and peripheral blood cells. This was confirmed by Horike et al,20 who showed that DLX5 is expressed monoallelic in these cells in normal individuals, but biallelic in patients with Rett syndrome. However, Schule et al,21 and recently Nakashima et al,22 argued that DLX5 was biallelic expressed, with highly variable expression of DLX5. So it is unclear whether DLX5 and DLX6 are under the influence of imprinting in the limb bud. Monoallelic expression would have important consequences for the pathogenesis of SHFM. As DLX5 would be expressed only from the maternal allele, only defects on the maternal chromosome 7 would cause SHFM. However, in Wieland et al11 it was explicitly stated that the deletion was paternal in origin, whereas in Weimer et al23 the translocated chromosome 7 was from maternal origin. Also in the family studied by Palmer et al24 both offspring of an affected male and of two affected females had SHFM, without a difference in severity of the phenotype. So it is unlikely that imprinting defects could be causative of the SHFM phenotype.
Deafness occurred in both our patients with a deletion and not in our patient with the inversion. Nonetheless, deafness has been reported not only in patients with a 7q21q22 deletion but also in patients with a balanced 7q rearrangement.1, 7 It is not known whether the same gene defect is responsible for both SHFM and deafness, or if two different genes in the SHFM1 region are involved.11, 25
In our third patient, PDD-NOS has been diagnosed. A relation between SHFM and autism has not been described previously. The occurrence of PDD-NOS might be explained by an imbalance or a disruption of a gene at the 7p22 break point. An imbalance was excluded by 244K oligo array-CGH. And because this region has never been identified in genome-wide association studies for autism,26, 27 a causative function of the distal break point in the PDD-NOS of our patient seems unlikely.
Of the cases summarized in Figure 3, we will discuss the cytogenetically well-characterized patient reported by Bernardini et al1 in more detail. This 5-year-old girl had a reciprocal interstitial translocation t(7;8)(q21q22;q23q24), with a paracentric inversion of 7q and a microdeletion of 7q21.13. The deleted segment included two genes, PFK1 and FZD1. Bernardini et al suggested that FZD1 could be a new candidate gene for SHFM1. FZD1 is a member of the ‘frizzled’ gene family, encoding a highly conserved Wnt receptor.28 It has been shown that the FZD1 protein is activated by WNT1 and WNT3A.29 Because Wnt1 and Wnt3A are known to interact with Dlx5 and Dlx6,30 Bernardini et al suggested that FZD1 is involved in limb development.1 FZD1 was deleted in our patients 1 and 2. However, in our third patient the inversion break point was located more than 4 Mb away from this gene.
Alternatively, Bernardini et al suggested that another, as yet unidentified, gene mapping to the region was involved in this patient's phenotype. FISH analysis disclosed that one of the break points of their paracentric inversion 7q22.1q31.2, about 5 Mb of the SHFM1 region, interrupted the CUTL1 gene. Experimental data have shown that Cutl1 is a member of the CCAAT displacement protein transcription factors family, involved in apical ectodermal ridge positioning and polarizing activities, and is regulated by Wnt pathway targets.31 In our patients, CUTL1 was not involved in the deletions of patients 1 and 2, and in patient 3 CUTL1 was located more than 5 Mb away from the inversion break point.
And finally, the inversion in Bernardini's patient was complicated by a translocation of a small 7q21.3 segment to chromosome 8. This translocated segment contained the three candidate genes, DLX5, DLX6 and DSS1. As probe RP11-15F5 was within the translocated segment, the proximal break point is at least 400 kb centromeric of the DLX genes. A comparable case was reported by Weimer et al23 describing two patients with split feet due to a complex translocation pattern involving chromosomes 2, 3 and 7. Among others, a part of chromosome 7q was translocated to chromosome 3, with the proximal break point just centromeric of DSS1 (100–400 kb) and thus about 400–700 kb centromeric of the DLX genes. It was suggested that the translocations in the patients of Bernardini et al1 and Weimer et al23 could have separated these genes from a control element. This is in line with our own observation and the studies in mice. On the basis of these well-defined cases and our patient 3, it is likely that the control element should be at least 800 kb centromeric of DLX5 and DLX6 (Figure 3).
In conclusion, the SHFM1 phenotype is most likely to be due to the simultaneously reduced expression of DLX5 and DLX6, comparable to the situation in SHFM4. This could be due to the concurrent deletions or true haploinsufficiency of these genes on one of the chromosome 7 pair. In addition, our results support the hypothesis that SHFM1 could also result from ‘functional haploinsufficiency’, a chromosomal rearrangement that physically separates the genes from their control element.
References
Bernardini L, Palka C, Ceccarini C et al: Complex rearrangement of chromosome 7q21.13–q22.1 confirms the ectrodactyly-deafness locus and suggests new candidate genes. Am J Med Genet A 2008; 146A: 238–244.
Basel D, Kilpatrick MW, Tsipouras P : The expanding panorama of split hand foot malformation. Am J Med Genet A 2006; 140A: 1359–1365.
Lo Iacono N, Mantero S, Chiarelli A et al: Regulation of Dlx5 and Dlx6 gene expression by p63 is involved in EEC and SHFM congenital limb defects. Development 2008; 135: 1377–1388.
de Mollerat XJ, Gurrieri F, Morgan CT et al: A genomic rearrangement resulting in a tandem duplication is associated with split hand-split foot malformation 3 (SHFM3) at 10q24. Hum Mol Genet 2003; 12: 1959–1971.
Ianakiev P, Kilpatrick MW, Toudjarska I, Basel D, Beighton P, Tsipouras P : Split-hand/split-foot malformation is caused by mutations in the p63 gene on 3q27. Am J Hum Genet 2000; 67: 59–66.
van Bokhoven H, Hamel BC, Bamshad M et al: p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet 2001; 69: 481–492.
Scherer SW, Poorkaj P, Massa H et al: Physical mapping of the split hand/split foot locus on chromosome 7 and implication in syndromic ectrodactyly. Hum Mol Genet 1994; 3: 1345–1354.
Scherer SW, Poorkaj P, Allen T et al: Fine mapping of the autosomal dominant split hand/split foot locus on chromosome 7, band q21.3–q22.1. Am J Hum Genet 1994; 55: 12–20.
Cobben JM, Verheij JBGM, Eisma WH et al: Bilateral split hand/foot malformation and inv(7)(p22q21.3). J Med Genet 1995; 32: 375–378.
Elliott AM, Evans JA : Genotype-phenotype correlations in mapped split hand foot malformation (SHFM) patients. Am J Med Genet A 2006; 140A: 1419–1427.
Wieland I, Muschke P, Jakubiczka S, Volleth M, Freigang B, Wieacker PF : Refinement of the deletion in 7q21.3 associated with split hand/foot malformation type 1 and Mondini dysplasia. J Med Genet 2004; 41: e54.
Robledo RF, Rajan L, Li X, Lufkin T : The Dlx5 and Dlx6 homeobox genes are essential for craniofacial, axial, and appendicular skeletal development. Genes Dev 2002; 16: 1089–1101.
Bongers EMHF, de Wijs IJ, Marcelis C, Hoefsloot LH, Knoers NVAM : Identification of entire LMX1B gene deletions in nail patella syndrome: evidence for haploinsufficiency as the main pathogenic mechanism underlying dominant inheritance in man. Eur J Hum Genet 2008; 16: 1240–1244.
Crackower MA, Scherer SW, Rommens JM et al: Characterization of the split hand/split foot malformation locus SHFM1 at 7q21.3–q22.1 and analysis of a candidate gene for its expression during limb development. Hum Mol Genet 1996; 5: 571–579.
Jantti J, Lahdenranta J, Olkkonen VM, Soderlund H, Keranen S : SEM1, a homologue of the split hand/split foot malformation candidate gene Dss1, regulates exocytosis and pseudohyphal differentiation in yeast. Proc Natl Acad Sci USA 1999; 96: 909–914.
Duijf PHG, van Bokhoven H, Brunner HG : Pathogenesis of split-hand/split-foot malformation. Hum Mol Genet 2003; 12: R51–R60.
Kobayashi K, Sinasac DS, Iijima M et al: The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet 1999; 22: 159–163.
Crackower MA, Sinasac DS, Xia J et al: Cloning and characterization of two cytoplasmic dynein intermediate chain genes in mouse and human. Genomics 1999; 55: 257–267.
Okita C, Meguro M, Hoshiya H, Haruta M, Sakamoto YK, Oshimura M : A new imprinted cluster on the human chromosome 7q21–q31, identified by human-mouse monochromosomal hybrids. Genomics 2003; 81: 556–559.
Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T : Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet 2005; 37: 31–40.
Schule B, Li HH, Fisch-Kohl C, Purmann C, Francke U : DLX5 and DLX6 expression is biallelic and not modulated by MeCP2 deficiency. Am J Hum Genet 2007; 81: 492–506.
Nakashima N, Yamagata T, Mori M, Kuwajima M, Suwa K, Momoi MY : Expression analysis and mutation detection of DLX5 and DLX6 in autism. Brain Dev 2009; doi:10.1016/j.braindev.2008.12.021.
Weimer J, Kiechle M, Wiedemann U et al: Delineation of a complex karyotypic rearrangement by microdissection and CGH in a family affected with split foot. J Med Genet 2000; 37: 442–445.
Palmer SE, Scherer SW, Kukolich M et al: Evidence for locus heterogeneity in human autosomal dominant split hand/split foot malformation. Am J Hum Genet 1994; 55: 21–26.
Tzschach A, Menzel C, Erdogan F et al: Characterization of a 16 Mb interstitial chromosome 7q21 deletion by tiling path array CGH. Am J Med Genet A 2007; 143A: 333–337.
Szatmari P, Paterson AD, Zwaigenbaum L et al: Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet 2007; 39: 319–328.
Yang MS, Gill M : A review of gene linkage, association and expression studies in autism and an assessment of convergent evidence. Int J Dev Neurosci 2007; 25: 69–85.
Bhanot P, Brink M, Samos CH et al: A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 1996; 382: 225–230.
Gazit A, Yaniv A, Bafico A et al: Human frizzled 1 interacts with transforming Wnts to transduce a TCF dependent transcriptional response. Oncogene 1999; 18: 5959–5966.
Riccomagno MM, Takada S, Epstein DJ : Wnt-dependent regulation of inner ear morphogenesis is balanced by the opposing and supporting roles of Shh. Genes Dev 2005; 19: 1612–1623.
Tavares AT, Tsukui T, Izpisua Belmonte JC : Evidence that members of the Cut/Cux/CDP family may be involved in AER positioning and polarizing activity during chick limb development. Development 2000; 127: 5133–5144.
Scherer SW, Cheung J, MacDonald JR et al: Human chromosome 7. DNA sequence and biology. Science 2003; 300: 767–772.
Acknowledgements
We are grateful to the patients and their parents and thank Mrs Jackie Senior for editing the paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
van Silfhout, A., van den Akker, P., Dijkhuizen, T. et al. Split hand/foot malformation due to chromosome 7q aberrations(SHFM1): additional support for functional haploinsufficiency as the causative mechanism. Eur J Hum Genet 17, 1432–1438 (2009). https://doi.org/10.1038/ejhg.2009.72
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2009.72
Keywords
This article is cited by
-
The molecular genetics of human appendicular skeleton
Molecular Genetics and Genomics (2022)
-
Phenotypic subregions within the split-hand/foot malformation 1 locus
Human Genetics (2016)
-
Deletions of exons with regulatory activity at the DYNC1I1 locus are associated with split-hand/split-foot malformation: array CGH screening of 134 unrelated families
Orphanet Journal of Rare Diseases (2014)
-
Split-hand/foot malformation - molecular cause and implications in genetic counseling
Journal of Applied Genetics (2014)
-
Balanced into array: genome-wide array analysis in 54 patients with an apparently balanced de novo chromosome rearrangement and a meta-analysis
European Journal of Human Genetics (2011)
