
Abstract
Autosomal recessive retinitis pigmentosa (ARRP) is a genetically heterogeneous disorder. ARRP could be associated with extraocular manifestations that define specific syndromes such as Usher syndrome (USH) characterized by retinal degeneration and congenital hearing loss (HL). The USH type II (USH2) associates RP and mild-to-moderate HL with preserved vestibular function. At least three genes USH2A, the very large G-protein-coupled receptor, GPR98, and DFNB31 are responsible for USH2 syndrome. Here, we report on the segregation of non-syndromic ARRP and USH2 syndrome in a consanguineous Tunisian family, which was previously used to define USH2B locus. With regard to the co-occurrence of these two different pathologies, clinical and genetic reanalysis of the extended family showed (i) phenotypic heterogeneity within USH2 patients and (ii) excluded linkage to USH2B locus. Indeed, linkage analysis disclosed the cosegregation of the USH2 phenotype with the USH2C locus markers, D5S428 and D5S618, whereas the ARRP perfectly segregates with PDE6B flanking markers D4S3360 and D4S2930. Molecular analysis revealed two new missense mutations, p.Y6044C and p.W807R, occurring in GPR98 and PDE6B genes, respectively. In conclusion, our results show that the USH2B locus at chromosome 3p23–24.2 does not exist, and we therefore withdraw this locus designation. The combination of molecular findings for GPR98 and PDE6B genes enable us to explain the phenotypic heterogeneity and particularly the severe ocular affection first observed in one USH2 patient. This report presents an illustration of how consanguinity could increase familial clustering of multiple hereditary diseases within the same family.
Similar content being viewed by others


Introduction
Hereditary retinal degenerations are a genetically and clinically heterogeneous group of disorders. The most common form of these disorders is retinitis pigmentosa (RP) affecting approximately 1 over 5000 individuals worldwide.1, 2 RP primarily affects the rod photoreceptors, whereas the function of the cone receptors is compromised as the disease progresses.3 RP may be inherited as an autosomal recessive, autosomal dominant or as an X-linked recessive trait. Autosomal recessive RP (ARRP) comprises 20–25% of cases of RP. To date, 40 loci have been implicated in non-syndromic RP, of which 30 genes are known, including PDE6B gene (http://www.sph.uth.tmc.edu/Retnet/disease.htm). This latter gene encodes the β-subunit of retinal rod cGMP phosphodiesterase, a key enzyme involved in phototransduction in rod photoreceptor cells.4 ARRP could also be associated with extraocular manifestations that define specific syndromes such as Usher syndrome (USH) (MIM276900), which represents the most common form of hereditary deafblindness in humans. It has a prevalence of ∼1/20 000 and represents 50% of the cases with deafblindness.5, 6, 7 USH is a group of autosomal recessive disorders characterized by RP with sensorineural hearing loss (HL) and variable vestibular function. Three clinically distinguishable USH types have been reported.8 People with USH type II (USH2) display congenital moderate-to-severe HL, RP and normal vestibular function. USH2 is genetically heterogeneous with at least three genes so far identified, USH2A, the very large G-protein-coupled receptor (GPCR), GPR98, and DFNB31.9, 10, 11
Using a consanguineous Tunisian family, we previously localized the USH2B locus to a 23-cM region on chromosome 3p23–24.2.12 Here, we report the segregation of missense alleles of GPR98 and PDE6B in the original and the single USH2B family described so far. Interestingly, the overlapping of both mutations underlies severe ocular affection. This report also presents an illustration of how consanguinity could increase familial clustering of multiple diseases within the same family.
Materials and methods
Clinical evaluation
Detailed clinical description of the USH2B family has been reported earlier.12, 13 Careful clinical re-examination of the USH2B family revealed the segregation of non-syndromic RP within another branch from the USH2B family. A complete historic interview enabled us to draw an extended pedigree from the family and to confirm the segregation of two different diseases, USH2 and non-syndromic ARRP, within the same previously reported USH2B family. A total of 21 additional members were currently ascertained including nine healthy individuals, six USH2 and six non-syndromic ARRP patients. All of them were evaluated for HL by pure-tone audiometry using air and bone conductions (frequencies ranging from 250 to 8000 Hz). Clinical histories were obtained from participating members to rule out obvious environmental causes of HL. Vestibular dysfunction was assessed by caloric tests in patients. All patients from the extended family underwent an ophthalmological examination including external eye examination, corrected visual acuity and funduscopic examination. Physical evaluations were undertaken to verify whether the RP was syndromic or non-syndromic. All USH2 family members from the prior studies along with the new ascertained ones were included in this study. The clinical diagnosis of USH2 or ARRP phenotype was established mainly on hearing evaluation and comparison of the onset, severity and progression of the retinal rod–cone dystrophy. Blood samples were collected from the new affected and unaffected family members.
Genotyping and linkage analysis
To confirm linkage to the USH2B locus, additional microsatellite markers were tested within the extended family. These markers were designed from the Généthon human linkage map14 and from the Marshfield chromosome-3 map (http://research.marshfieldclinic.org/genetics). Furthermore, a genome-wide screening was performed with 400 microsatellite markers distributed at average intervals of 10 cM (ABI Prism Linkage Mapping Set 2, Applied Biosystems). Fluorescently labeled alleles were analyzed on an ABI PRISM 3100-Avant automated DNA sequencer (Applied Biosystems, USA). Genetic analysis of non-syndromic ARRP loci was undertaken using fluorescently labeled primers surrounding microsatellite repeats at known RP loci selected from Marshfield chromosome maps (Table 1). Two-point linkage analyses were performed using the FASTLINK version of MLINK from the LINKAGE Program Package.15, 16 Maximum LOD scores were calculated using ILINK. USH2 syndrome was analyzed as a fully penetrant trait with an estimated frequency of 10−3 for the disease-causing allele. Recombination frequencies were assumed to be equal for male and female individuals, and allele frequencies of linkage markers were assumed to be equal.
Mutation screening
Candidate genes in the USH2B interval on chromosome 3p23–24.2 were identified either using the UCSC Genome browser (http://www.genome.ucsc.edu/) or suggested by collaborators. Primers used for PCR amplification and subsequent sequencing of USH2B candidate genes were designed from the flanking region of each exon using Primer3 web utility (http://www.frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi/). We also analyzed all the 90 exons of the GPR98 gene (GeneID: 84059) and the 22 exons of PDE6B gene (GeneID: 5158). Primers were synthesized from the consensus intronic sequences except for sets of exonic primers designed to amplify the overlapping parts of exons. To study the c.18131A>G change occurring in exon 85 of the GPR98 gene, we screened 95 genetically unrelated controls using a PCR/RFLP approach: exon 85 of the GPR98 gene was amplified with intronic primers resulting in a product of 325 bp. Restriction analysis with Hpy8I of the wild-type allele results in two fragments of 265 and 60 bp, whereas the mutated allele is cleaved into three fragments of 239, 60 and 26 bp. The mutation introduces a novel Hpy8I site GTNNAC.
Results
Earlier, we had mapped the USH2B gene to a region on chromosome 3p23–24 using a consanguineous Tunisian family named ‘Us’.12 Although the corresponding region was large (23 cM), attractive interesting genes were selected as positional candidates in the USH2B interval (TMIE;17 SLC26A6 (MIM610068), NBC3 (MIM603353) and CELSR3 (MIM604264)). However, no disease-associated alteration was identified in any of them.
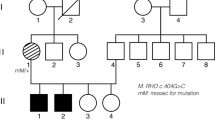
In parallel, we had the opportunity to extend the ‘Us’ pedigree by studying 21 additional healthy and affected individuals as well as a new nuclear-related branch affected by non-syndromic ARRP. The genotyping of the extended ‘Us’ family with additional microsatellite markers flanking the USH2B interval did not show any homozygous region shared between all USH2 patients, and some of them still show a heterozygous USH2B haplotype (Figure 1; individuals VI-8, VI-9 and VI-10). Moreover, the genotyping of newborn affected USH2 patients VII-10, VII-11 and VII-12 failed to show any linkage and excluded the involvement of the USH2B locus in the family (data not shown). Given the concomitant segregation of USH2 and non-syndromic ARRP in the same family, we undertook a clinical reanalysis on the extended family. All patients and healthy family members in the prior study were included in this study. Clinical examination showed moderate-to-severe non-progressive congenital HL in all USH2-affected members (Table 2 and Figure 2). Vestibular function appeared to be normal in affected individuals and clinical evaluation suggested no skin or renal anomalies. Ophthalmologic examination displayed a progressive night blindness and constriction of the peripheral visual field, with mild-to-severe loss of central vision (Supplementary Figure 1). Compared with younger patients, older subjects showed evidence of deteriorated cone function that indicates the progressive nature of the retinal dystrophy. All patients underwent dilated indirect fundoscopy. Recorded changes included progressive dystrophy of the retina in the retinal periphery, with or without macular involvement, with intraretinal pigment migration, attenuation of the retinal blood vessels and some degree of pallor of the optic disc (Figure 3c). Moreover, in patient VI-10, a 39-year-old woman, fundus photographs showed a more severe RP that had progressed and covered almost the whole retina reaching the center (Figure 3c). When interviewing her relatives, they confirmed the early appearance of night blindness in that woman starting from the beginning of her first decade. Interestingly, this early severe ocular defect was also observed in all patients affected by non-syndromic ARRP. Indeed, those patients share the precocity of the RP onset, which started early at the age of 2 years and subsequently progressed severely until invading the whole retina. We therefore suspected in the ‘Us’ family, the segregation of two different pathologies non-syndromic ARRP and the USH2 syndrome, which have overlapped in patient VI-10 explaining the severity of her retinal dystrophy.

Pedigree and segregation analysis of the extended ‘Us’ family. Haplotypes are shown only for members relevant to the study and represented as columns of numbers, with the ancestral disease-associated haplotype boxed. Individual VI-5 provides a recombination event indicated by an arrow.  ,
,  =male and female individuals affected with USH2 syndrome;
=male and female individuals affected with USH2 syndrome;  ,
,  =male and female individuals affected with ARRP; ▪, •=male and female individuals affected with both USH2 syndrome and ARRP.
=male and female individuals affected with ARRP; ▪, •=male and female individuals affected with both USH2 syndrome and ARRP.

Audiograms from USH2 patients and one healthy individual from ‘Us’ family. The hearing threshold values were assessed at sound frequencies 250, 500, 1000, 2000, 4000 and 8000 Hz.

(a) Pedigree and haplotype of family ‘Us’ for VLGR1 and PDE6B chromosomal regions. Haplotype analysis revealed that the index patient VI-10 is homozygous for microsatellite markers flanking both the VLGR1 and the PDE6B loci. (b) Mutation analysis revealed two new missense mutations, c.18131G>A and c.2419T>A, occurring in VLGR1 and PDE6B genes, respectively. The parents V-1 and V-2 are double heterozygous for both mutations, and patients showing either ARRP (VI-3) or USH2 (VI-5) alone are homozygous only for c.2419T>A or c.18131A>G, respectively. However, the index patient VI-10 is homozygous for both alterations. (c) Fundus photographs of healthy father, affected individuals presenting with ARRP and USH2 and both forms of retinal dystrophies. There are changes typical of retinitis pigmentosa including attenuation of retinal arteries and bone-spicule pigment deposits in the mid-periphery of the retina. In patient VI-10, note the severe RP that has progressed and covered almost the whole retina reaching the center. (d) Restriction analysis with Hpy8I. Exon 85 of the GPR98 gene was amplified with intronic primers resulting in a product of 325 bp. Digestion of the wild-type allele results in two fragments of 265 and 60 bp. Although the mutated allele is cleaved into three fragments of 239, 60 and 26 bp, the mutation introduces a novel Hpy8I site GTNNAC.
Genetic findings excluding the USH2B locus, along with the phenotypic heterogeneity observed between the USH2 patients, prompted us to reanalyze the extended family with regard to the concomitant segregation of two different pathologies that is, non-syndromic ARRP and USH2 syndrome. We therefore considered only those patients with both HL and RP as USH2 and all the others as normal but those with RP alone were excluded. We undertook a screening of the whole genome using 400 microsatellite markers at intervals of 10 cM. Surprisingly, a linkage was detected only with the microsatellite marker D5S428 on chromosome 5, corresponding to the USH2C locus. The addition of other markers within the same region and haplotype analysis confirmed the linkage (Figure 1) and disclosed a recombination event occurring in affected individual VI-5 between D5S641 and D5S428. A maximum two-point LOD score of 4.1 was obtained with microsatellite marker D5S618 at a recombination fraction θ=0.00 (Table 3). None of RP patients showed the pathologic USH2C haplotype in two copies, ruling out the involvement of this locus in both phenotypes. We performed, thereby, a genetic analysis of the known ARRP genes using the corresponding microsatellite markers (Table 1). Linkage analysis showed cosegregation of the RP branch with the telomere of chromosome 4 harboring the PDE6B gene. All ARRP patients were homozygous for both D4S3360 and D4S2936 markers (Figure 3a). Molecular analysis identified a new substitution c.2419T>A, in exon 21 of PDE6B gene, that segregates perfectly with the ARRP phenotype in the family. This change results in the substitution of an evolutionarily conserved tryptophan with a polar arginine at position 807 (Figure 4a).

Sequence alignments of GPR98 (a) and PED6B (b) proteins from vertebrate species for which the sequence was present in Refseq database (accession numbers are given after species common name). The mutated residues are highlighted in pink in online version.
Sequencing of the GPR98 coding region in an affected member VI-11 revealed homozygosity for a missense mutation c.18131A>G occurring in exon 85 resulting in the substitution of the residue tyrosine at position 6044 by a cysteine (p.Y6044C). This change perfectly segregates with the USH2 phenotype. Indeed, all patients were homozygous for the c.18131A>G allele and healthy individuals were either heterozygous or homozygous for the wild-type allele. The two parents V-1 and V-2, although aged 82 and 65 years, respectively, did not show any alteration of the retinal pigmented epithelium or hearing problems despite being double heterozygous for the c.2419T>A and the c.18131A>G mutations (Figure 3a and b). Using a PCR/RFLP approach, we did not find this mutation in 95 genetically unrelated controls. In addition, the c.18131A>G mutation was tested in 24 American USH2 families unlinked to USH2A locus and was found to be in a heterozygous state in one USH2 family. This mutation results in the substitution of a highly conserved tyrosine residue at position 6044 with a cysteine at the second extracellular loop (ECL2) of the GPR98 (Figure 4b). This newly introduced cysteine residue may disrupt the interloop disulfide bridge between ECL1 and ECL2 leading to an improperly folded loop and consequently to a non-functional receptor. Sequence analysis also identified two new sequence variants, c.16031G>A and c.16279G>A, in the coding exons 74 and 76, respectively, and one nucleotide change (T>C) in intron 25. They were considered as polymorphisms as they have been detected in several unaffected individuals (unpublished data). Interestingly, the combination of GPR98 and PDE6B molecular findings disclosed a double homozygosity involving both genes in the same patient VI-10, which would be expected to be responsible for her severe ocular phenotype (Figure 3c).
Discussion
This study deals with the segregation of two different forms of hereditary retinal degenerations including non-syndromic ARRP and USH2 syndrome within the same consanguineous Tunisian family. Such a familial aggregation of hereditary diseases has been well described in different populations, in particular those with a high consanguinity rate.18, 19 Indeed, the Tunisian population consists of a mosaic of communities (Phoenicians, Roman, Berber, Arab, Ottoman, etc.). This heterogeneity along with geographic isolation, social traditions and parental involvement in mate selection all contribute to increase consanguinity in these regions. In addition to this aggregation, the consanguinity also seems to be involved in concentrating some diseases, increasing their prevalence in the corresponding population. In fact, although USH2 syndrome seems to be the most common type of USH in western European population,9 this form seems to be much less frequent in Tunisian population. It is likely that in certain consanguineous populations, such as Tunisian population, specific types of USH could be relatively frequent because of founder effects.20, 21, 22 Alternatively, this low frequency of the USH2 type could also be attributed to an under diagnosis of this clinical type justified by its mild ocular and auditive affection.
We have previously localized the USH2B gene to a 23-cM region on chromosome 3p23–24.2 using the ‘Us’ Tunisian family.12 As far as we extended the study on the large family, genetic analysis clearly excluded the involvement of a gene in the USH2B region and thus we withdrew USH2B locus designation. Moreover, clinical findings showed phenotypic heterogeneity in USH2 patients particularly in the ocular affection with regard to the severity and the RP onset. Typically, RP leads to the development of night blindness and loss of peripheral vision. Progressive retinopathy, clinically indistinguishable from classic RP, typically develops in the childhood to adolescent years in all types of USH.23, 24, 25 However, instances of late-onset RP in cases of USH2 syndrome have been reported.26 Within our family, we noted late-onset RP in the majority of USH2 patients, with the exception of patient VI-10 presenting with an early-onset RP that has already invaded almost the whole retina. Interestingly, this later severe and early-onset phenotype was also present in all ARRP patients, raising the possibility of two overlapping pathologies in patient VI-10.
Genetic and molecular analysis revealed the coexistence of mutations affecting two different genes GPR98 and PDE6B, ruling out the involvement of the same gene in both forms of hereditary retinal degeneration. In fact, with the exception of specific USH2A mutations, no change in any of the Usher genes has been implicated thus far as a cause of RP without HL. Indeed, besides as USH2, some mutations in the USH2A gene are manifested as atypical USH26 or as non-syndromic recessive RP27, 28 without hearing or balance deterioration over time in these patients. In addition, each of these USH2A mutations occurred separately in an individual family. Thus, it remains still unknown whether disease mechanisms and dynamics of USH are the same as those underlying non-syndromic RP.
We discovered a missense mutation in exon 21 of PDE6B gene at position 2419 where thymidine is replaced by adenine resulting in the substitution of polar arginine with tryptophan. Although the mutation lies outside the catalytic domain of the enzyme, it occurs at an evolutionarily conserved position. It has been postulated through studies on murine and canine models that defective functioning of the phosphodiesterase is detrimental for overall health of rod photoreceptors. Malfunctioning of PDE results in elevated levels of cGMP. Apoptosis is induced due to excess cations flowing in the rod cells leading to retinal degeneration.29, 30
Molecular analysis of the coding region of the GPR98 gene, a member of the GPCR family, showed a homozygous missense mutation c.18131A>G occurring in exon 85 and resulting in the substitution of a highly conserved tyrosine with a cysteine at position 6044 in the ECL2 of the GPR98 receptor. This mutation was also found in a heterozygous state in one USH2 family unlinked to USH2A locus. Indeed, the majority of family A and family B GPCR contains a conserved pair of extracellular cysteine residues linking ECL1 and ECL2 through a disulfide bond. Numerous functional analyses of GPCR mutants where cysteine residues were replaced by other amino acids have shown that this disulfide bond may be critical for receptor signaling.31, 32, 33 The structural consequences of the presence of additional cysteine residues within the ECL seem to be complex. It is likely that this newly introduced residue cysteine 6044 may offer alternatives in disulfide bond formation that compete and disrupt the interloop disulfide bridge between ECL1 and ECL2, leading to an improperly folded loop and consequently to a non-functional receptor. Alternatively, this missense mutation may alter the intracellular trafficking of the receptor protein. In fact, more than 80% of small inframe deletion/insertion and missense mutations lead to improperly folded proteins retained intracellularly by the quality control machinery of the endoplasmic reticulum.34, 35
Disease-causing mutations of the highly conserved disulfide bond connecting ECL1 and ECL2 have been identified in many GPCRs, including AVPR2,36 RHO37, 38 and MC2R.39 About 7% of all missense mutations in GPCR are substitutions with cysteine residues found in the putative N-terminal portion and ECL1-3.40 Indeed, these extracellular loops of GPCR frequently contain binding sites for peptide ligands. However, the mechanism of receptor activation following ligand binding and the influence of the extracellular loops in aspects of receptor function are poorly understood.
Mutations so far reported in the GPR98 gene are mostly nonsense mutations occurring either in the large N-terminal ectodomain or in the C-terminal region resulting in a dramatically truncated protein.10, 41 This is the first report on the occurrence of pathogenic missense mutations in the extracellular domain of GPR98, suggesting that this domain could fulfil a range of important roles with respect to GPCR signaling including agonist binding and/or receptor activation. Moreover, unlike the complement factor 5a receptor, C5aR, in which the ECL2 plays a negative regulating role in the receptor activation, ECL2 of GPR98 seems to have a positive regulator effect. In fact, this amino-acid substitution has disrupted the receptor function instead of generating a ligand-independent activity receptor. Thus, we could suggest a two-site model for ligand binding to GPR98 supporting other groups, proposing that along with the amino terminus part, the extracellular loops could influence the high-affinity binding of the GPR98 ligand.
Recent studies have reported that GPR98 is a component of the ankle link complex,42, 43which is one among four distinct types of links that connect stereocilia forming the sensorineural cell hair bundle. This interstereocilial complex, involving also Usherin and likely protocadherin-15,44, 45 is required for normal development of auditory hair bundles explaining anomalies observed in USH2 patients.
References
Bunker CH, Berson EL, Bromley WC, Hayes RP, Roderick TH : Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol 1984; 97: 357–365.
Bundey S, Crews SJ : A study of retinitis pigmentosa in the city of Birmingham. J Med Genet 1986; 23: 188.
Bird AC : Retinal photoreceptor dystrophies LI. Edward Jackson Memorial Lecture. Am J Ophthalmol 1995; 119: 543–562.
Bayes M, Giordano M, Balcells S et al: Homozygous tandem duplication within the gene encoding the β-subunit of rod phosphodiesterase as a cause for autosomal recessive retinitis pigmentosa. Hum Mutat 1995; 5: 228–234.
Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, Nance WE : Genetic epidemiological studies of early-onset deafness in the US school-age population. Am J Med Genet 1993; 46: 486–491.
Rosenberg T, Haim M, Hauch AM, Parving A : The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin Genet 1997; 51: 314–321.
Spandau UH, Jonas JB, Gass A : Progressive visual loss in optic nerve hypoplasia and bilateral microdiscs. Arch Neurol 2002; 59: 1829–1830.
Keats BJ, Corey DP : The usher syndromes. Am J Med Genet 1999; 89: 158–166.
Eudy JD, Weston MD, Yao S et al: Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science 1998; 280: 1753–1757.
Weston MD, Luijendijk MW, Humphrey KD, Möller C, Kimberling WJ : Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet 2004; 74: 357–366.
Ebermann I, Scholl HP, Charbel Issa P et al: A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet 2007; 121: 203–211.
Hmani M, Ghorbel A, Boulila-Elgaied A et al: A novel locus for Usher syndrome type II, USH2B, maps to chromosome 3 at p23–24.2. Eur J Hum Genet 1999; 7: 363–367.
Hmani-Aifa M, Ben Arab S, Kharrat K et al: Distinctive audiometric features between USH2A and USH2B subtypes of Usher syndrome. J Med Genet 2002; 39: 281–283.
Dib C, Fauré S, Fizames C et al: A comprehensive genetic map of the human genome based on 5264 microsatellites. Nature 1996; 380: 152–154.
Lathrop GM, Lalouel JM, Julier C, Ott J : Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 1984; 81: 3443–3446.
Schaffer AA, Gupta SK, Shriram K, Cottingham Jr RW : Avoiding recomputation in linkage analysis. Hum Hered 1994; 44: 225–237.
Naz S, Giguere CM, Kohrman DC et al: Mutations in a novel gene, TMIE, are associated with hearing loss linked to the DFNB6 locus. Am J Hum Genet 2002; 71: 632–636.
Farag TI, Teebi AS : Genetic disorders among the Bedouins; in Teebi AS Farag TI (eds): Genetic Disorders among Arab Populations. New York: Oxford University Press, 1997, pp 375–410.
Becker S, Al Halees Z : First-cousin mating and congenital heart disease in Saudi Arabia. Community Genet 1999; 2: 69–73.
Keats BJ, Nouri N, Pelias MZ, Deininger PL, Litt M : Tightly linked flanking microsatellite markers for the Usher syndrome type I locus on the short arm of chromosome 11. Am J Hum Genet 1994; 54: 681–686.
Pakarinen L, Karjalainen S, Simola KO, Laippala P, Kaitalo H : Usher's syndrome type 3 in Finland. Laryngoscope 1995; 105: 613–617.
Ness SL, Ben-Yosef T, Bar-Lev A : Genetic homogeneity and phenotypic variability among Ashkenazi Jews with Usher syndrome type III. J Med Genet 2003; 40: 767–772.
Fishman GA, Kumar A, Joseph ME, Torok N, Anderson RJ : Usher′s syndrome. Ophthalmic and neuro-otologic findings suggesting genetic heterogeneity. Arch Ophthalmol 1983; 101: 1367–1374.
Smith RJ, Berlin CI, Hejtmancik JF et al: Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am J Med Genet 1994; 50: 32–38.
Iannaccone A : Usher syndrome: correlation between visual field size and maximal ERG response b-wave amplitude. Adv Exp Med Biol 2003; 533: 123–131.
Liu XZ, Hope C, Liang CY et al: A mutation (2314delG) in the Usher syndrome type IIA gene: high prevalence and phenotypic variation. Am J Hum Genet 1999; 64: 1221–1225.
Rivolta C, Sweklo EA, Berson EL, Dryja TP : Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet 2000; 66: 1975–1978.
Bernal S, Ayuso C, Antiñolo G et al: Mutations in USH2A in Spanish patients with autosomal recessive retinitis pigmentosa: high prevalence and phenotypic variation. J Med Genet 2003; 40: 1–5.
Dryja TP, David ER, Sherleen HC, Eliot LB : Frequency of Mutations in the Gene Encoding the α Subunit of Rod cGMP-Phosphodiesterase in Autosomal Recessive Retinitis Pigmentosa. Invest Ophthalmol Vis Sci 1999; 40: 1859–1865.
Takahashi M, Miyoshi H, Verma IM, Gage FH : Rescue from photoreceptor degeneration in the rd mouse by human immunodeficiency virus vector-mediated gene transfer. J Virol 1999; 73: 7812–7816.
Kosugi S, Ban T, Akamizu T, Kohn LD : Role of cysteine residues in the extracellular domain and exoplasmic loops of the transmembrane domain of the TSH receptor: effect of mutation to serine on TSH receptor activity and response to thyroid stimulating autoantibodies. Biochem Biophys Res Commun 1992; 189: 1754–1762.
Savarese TM, Wang CD, Fraser CM : Site-directed mutagenesis of the rat m1 muscarinic acetylcholine receptor. Role of conserved cysteines in receptor function. J Biol Chem 1992; 267: 11439–11448.
Cook JV, Eidne KA : An intramolecular disulfide bond between conserved extracellular cysteines in the gonadotropin-releasing hormone receptor is essential for binding and activation. Endocrinology 1997; 138: 2800–2806.
Leanos-Miranda A, Janovick JA, Conn PM : Receptor-misrouting: an unexpectedly prevalent and rescuable etiology in gonadotropin-releasing hormone receptor-mediated hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2002; 87: 4825–4828.
Schülein R : The early stages of the intracellular transport of membrane proteins: clinical and pharmacological implications. Rev Physiol Biochem Pharmacol 2004; 151: 45–91.
Bichet DG, Birnbaumer M, Lonergan M, Arthus MF, Rosenthal W, Goodyer P : Nature and recurrence of AVPR2 mutations in X-linked nephrogenic diabetes insipidus. Am J Hum Genet 1994; 55: 278–286.
Al-Maghtheh M, Gregory C, Inglehearn C, Hardcastle A, Bhattacharya S : Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Hum Mutat 1993; 2: 249–255.
Fuchs S, Kranich H, Denton MJ, Zrenner E, Bhattacharya SS, Humphries P : Three novel rhodopsin mutations (C110F, L131P, A164V) in patients with autosomal dominant retinitis pigmentosa. Hum Mol Genet 1994; 3: 1203.
Naville D, Barjhoux L, Jaillard C, Faury D, Despert F, Esteva B : Demonstration by transfection studies that mutations in the adrenocorticotropin receptor gene are one cause of the hereditary syndrome of glucocorticoid deficiency. J Clin Endocrinol Metab 1996; 81: 1442–1448.
Schöneberg T, Schulz A, Gudermann T : The structural basis of G-protein-coupled receptor function and dysfunction in human diseases. Rev Physiol Biochem Pharmacol 2002; 144: 143–227.
Nakayama J, Fu YH, Clark AM, Nakahara S, Hamano K, Iwasaki N : A nonsense mutation of the MASS1 gene in a family with febrile and afebrile seizures. Ann Neurol 2002; 52: 654–657.
McGee J, Goodyear RJ, McMillan DR et al: The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J Neurosci 2006; 26: 6543–6553.
Goodyear RJ, McGee J, Weston MD et al: Identification of the ankle link antigen as the Usher 2c protein, VLGR1, a protein required for cochlear hair bundle development. Assoc Res Otolaryngol 2006; 29: 48.
Adato A, Lefèvre G, Delprat B et al: Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum Mol Genet 2005; 14: 3921–3932.
Ahmed ZM, Goodyear R, Riazuddin S et al: The tip-link antigen, a protein associated with the transduction complex of sensory hair cells, is protocadherin-15. J Neurosci 2006; 26: 7022–7034.
Acknowledgements
We thank the family members for their invaluable participation and cooperation. We are thankful to Pr Saber Masmoudi for his critical reading of the paper. We also thank Salma ben Salem for her help in table and figure perfection. This study was supported by Ministère de l’Enseignement Supérieur, de la Recherche Scientifique et de la Technologie, Tunisia, and the European Commission FP6 Integrated Project EUROHEAR, LSHG-CT-20054-512063. Support for WJ Kimberling is from Foundation Fighting Blindness (BR-GE-0606-0343) and the National Institutes of Deafness and Communication Disorders (P01 DC01813).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Hmani-Aifa, M., Benzina, Z., Zulfiqar, F. et al. Identification of two new mutations in the GPR98 and the PDE6B genes segregating in a Tunisian family. Eur J Hum Genet 17, 474–482 (2009). https://doi.org/10.1038/ejhg.2008.167
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.167
Keywords
This article is cited by
-
A novel homozygous missense substitution p.Thr313Ile in the PDE6B gene underlies autosomal recessive retinitis pigmentosa in a consanguineous Pakistani family
BMC Ophthalmology (2023)
-
The genetics of rod-cone dystrophy in Arab countries: a systematic review
European Journal of Human Genetics (2021)
-
Natural models for retinitis pigmentosa: progressive retinal atrophy in dog breeds
Human Genetics (2019)
-
A novel homozygous variant of GPR98 causes usher syndrome type IIC in a consanguineous Chinese family by next generation sequencing
BMC Medical Genetics (2018)
-
Coexisting variants in OSTM1 and MANEAL cause a complex neurodegenerative disorder with NBIA-like brain abnormalities
European Journal of Human Genetics (2017)
