
Abstract
MEF2C haploinsufficiency syndrome is an emerging neurodevelopmental disorder associated with intellectual disability, autistic features, epilepsy, and abnormal movements. We report 16 new patients with MEF2C haploinsufficiency, including the oldest reported patient with MEF2C deletion at 5q14.3. We detail the neurobehavioral phenotype, epilepsy, and abnormal movements, and compare our subjects with those previously reported in the literature. We also investigate Mef2c expression in the developing mouse forebrain. A spectrum of neurofunctional deficits emerges, with hyperkinesis a consistent finding. Epilepsy varied from absent to severe, and included intractable myoclonic seizures and infantile spasms. Subjects with partial MEF2C deletion were statistically less likely to have epilepsy. Finally, we confirm that Mef2c is present both in dorsal primary neuroblasts and ventral gamma-aminobutyric acid(GABA)ergic interneurons in the forebrain of the developing mouse. Given interactions with several key neurodevelopmental genes such as ARX, FMR1, MECP2, and TBR1, it appears that MEF2C plays a role in several developmental stages of both dorsal and ventral neuronal cell types.
Similar content being viewed by others

Introduction
A number of patient series have described the major symptoms of haploinsufficiency of MEF2C at 5q14.3 (OMIM No. 613443) [1–8]. These include epilepsy, intellectual disability (ID), and autistic features such as absent speech, impaired social reciprocity, and stereotypical movements [9]. The details and variability of the major features, particularly the epilepsy, in MEF2C haploinsufficiency have not been systematically determined. Children with MEF2C mutations also have abnormal movement patterns, but video examples of these movements and their evolution across the lifespan have not been published. Finally, although patients with MEF2C haploinsufficiency have neurobehavioral deficits, there has not been a broad survey of neurologic function in a large number of these patients to date. Therefore, key aspects of this disorder remain incompletely characterized.
MEF2C is a transcription factor with changing expression patterns during brain development that correlate with roles in both early neuroprogenitor development [10, 11] and later during neuronal maturation [12–14]. There are three alternative exons capable of generating six Mef2c isoforms in the mouse, and the Mef2c α1β isoforms are expressed in neurons [15]. Complete loss of Mef2c is embryonic lethal, but conditional knockouts have demonstrated a role for Mef2c in both gamma-aminobutyric acid (GABA)ergic and glutaminergic pyramidal neuron migration [16], and in maintenance of synapse stability and function [17]. At early embryonic periods, Mef2c is a subplate and cortical plate marker [18], and postnatally, it is present throughout the cortical plate.
The expression of Mef2c has been monitored in a number of developmental gene knockout models. Mef2c expression is reduced in the Tbr1-knockout mouse, indicating that Mef2c plays a role in Tbr1 modulation of cortical layering [18]. Mef2c expression in the mouse forebrain is also reduced in the absence of Arx [19] and Dlx1/2 [20] suggesting that ARX and MEF2C are members of a transcriptional regulatory network in GABAergic interneurons. Additionally, the expression of two other important neurodevelopmental genes—CDKL5 and MECP2—is altered in children with mutations in MEF2C [7]. MEF2C plays a role in forebrain development—both dorsally and ventrally—but knowledge about its specific role in multiple neuronal developmental pathways remains incomplete.
We seek to clarify several of these outstanding issues by reporting 16 new subjects with MEF2C haploinsufficiency syndrome and detailing their neurologic phenotype. We report epilepsy subtypes, response to treatment and outcome, illustrate a consistent hyperkinetic movement disorder with video, and detail the extent of neurofunctional deficits in our subjects. We present our data in the context of the 27 MEF2C haploinsufficiency subjects in the published literature and where possible identify common and discordant features from this combined expanded cohort. To further clarify the potential mechanisms of the neurodevelopmental disorder in MEF2C haploinsufficient individuals, we re-demonstrate that Mef2c is present in both neuroblasts and GABAergic inhibitory interneurons in the forebrain of the developing mouse, with significant expression in GABAergic interneurons at embryonic day 14.5. Then, we perform a meta-analysis on publicly available data on the interactions of Mef2c with the neurodevelopmental genes Arx, Dlx1/2, Mecp2, and Tbr1. From these data, we conclude that MEF2C plays a role in multiple neuronal developmental pathways, and that the clinical phenotype likely reflects abnormalities of both dorsal and ventral forebrain development.
Methods
Patients
Subjects were identified through the Infantile Spasms Registry & Genetic Studies (ARP), search of a database of clinically obtained array CGH (aCGH) results at Signature Genomic Laboratories (LGS), or review of positive aCGH findings at collaborating pediatric neurology, genetics, and neurogenetics clinics. Retrospective records from 16 patients with MEF2C haploinsufficiency were reviewed, including EEG data, brain imaging, and physician descriptions of abnormal movements. Care was taken to ensure that non-epileptic movements were appropriately documented as such by the patient’s primary neurologist. Video of behavior and movements was reviewed in eight of the subjects. Movements were classified using standardized methods [21, 22]. Neurobehavioral and neurofunctional deficits were reviewed in 14 of the 16 families by telephone, or by using available records if parents were not available. The study was approved by the institutional review boards of Washington University, University of Rochester Medical Center, Seattle Children’s Hospital, and IRB-Spokane, including written consent to publish photographs and video.
Statistical analysis
t test comparing deletion size (partial or complete MEF2C deletion) and the presence of epilepsy, abnormal movements, or specific neurofunctional deficits was performed using R version 2.13.1.
Microarray-based comparative genomic hybridization (aCGH)
Microarray analysis was performed on genomic DNA between subjects LR11-305, LR11-306, LR11-307, LR11-308, and LR11-309 on a custom 105K-feature whole genomic microarray (Agilent Technologies, Santa Clara, CA, USA) as previously described [23]. Other subjects were assayed on a variety of clinically available platforms including Illumina BeadChip 6.0 (LR11-312), Illumina HumanQuad610 BeadChip SNP array (LR11-387), Affymetrix Whole-Genome 2.7M SNP array (LR11-325), GeneDx “GenomeDx” v1.0 oligo array (LR11-388), a custom Agilent oligo array with 40K features (LR12-031), 44K oligo array (LR12-013 and LR12-275), 180K oligo array (LR12-022), and 244K oligo array (LR12-021) according to the manufacturer instructions. Chromosome 5q14.3 deletions detected by aCGH were visualized with metaphase fluorescence in situ hybridization (FISH) using one or more BAC clones located within the abnormal regions as determined by aCGH, according to standard clinical protocols. Confirmatory FISH was not performed in subject LR11-325 with a large 6-Mb deletion. The deletion in subject LR11-388 was confirmed by quantitative PCR (qPCR) for exon 45 of GPR98. All studies were obtained as part of routine clinical evaluations, except for IS09-024, who was studied on an Affymetrix SNP array as part of a research protocol.
Animal assurance
All experiments were approved by and carried out in accordance with the Animal Care and Use Committee of the Children’s Hospital of Philadelphia. All mice were housed in a room with 12-h light–dark cycle and had continuous access to food and water.
Mef2c staining methods
The immunohistochemical experiments were performed on either C57/Bl6 mice or Dlx5/6 cre-reporter mice (Dlx5/6CRE-ires-GFP) that were on a C57/Bl6 background. Timed breeding between C57/Bl6 mice was performed, and embryos and pups were harvested at embryonic day 14.5 (e14.5). The brains were dissected out of the embryos and fixed in 4 % paraformaldehyde for 4–8 h. For older animals of P14 and adult ages (>P45), the animals were transcardially perfused, and then the brains were post-fixed overnight. The brains at all ages were cut on a cryostat at 14–18 μ. Sections were processed for immunohistochemistry as previously published [24]. For Mef2c staining, goat anti-Mef2c antibody (Santa Cruz Cat No. sc-13268 and Lot No. K2009) was used. The antibody (1:1,000 dilution in 10 % donkey serum) was incubated overnight at 4 °C. After three washes in PBS, a biotinylated (anti-goat) secondary antibody (1:2,000) dilution was applied for 30 min, followed by an avidin-conjugated to Cy3 fluorophore (1:500 dilution), both at room temperature. Tissue was visualized using a Leica DMR microscope (Leica Microsystems, Bannockburn, IL, USA) equipped with epifluorescence and light microscopy. Images were acquired on an Orca digital camera (Hamamatsu, Hamamatsu City, Japan) and processed with Image-Pro software (Media Cybernetics, Bethesda, MD, USA). White balance, auto contrast, and stitching of images were performed with Adobe Photoshop (Adobe Systems).
Gene expression arrays
The methods for the microarray data were published previously and are publicly available via the NIH neuroscience microarray consortium (http://np2.ctrl.ucla.edu/np2/home.do). Briefly, a Dlx5/6 reporter mouse line with GFP expression in migrating interneurons was used. Fluorescence-activated cell sorting (FACS) of GFP+ cells from the ganglionic eminence (GE) and cortex was performed. The array data were processed in Partek, and a false discovery rate (FDR) calculation was performed using a standard p value of 0.01. For this study, the Mef2c probes were queried for presence in migrating interneurons (Cortex+) versus non-interneurons (Cortex−) and both compared to GFP+ cells in the GE (GE+).
Analysis of publicly available gene expression array data
Data from Arx and Dlx1/2 knockout experiments were obtained by reviewing the published papers [19, 25, 26] and querying the relative expression and p values for the Mef2c probes in the available source arrays (see Online resources). We also reviewed the publicly available data from the Tbr1 knockout mouse [18] and mouse models of Mecp2 under- and over-expression [27].
Results
Patients
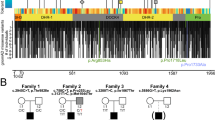
The key phenotype and genotype characteristics of the 16 subjects are shown in Table 1. They include 15 subjects with deletions of 5q14.3 containing MEF2C and one patient with a novel frameshift mutation in MEF2C. The genomic breakpoints of the subjects with 5q14.3 deletions are illustrated in Fig. 1.

Genomic data showing deletions (red) of 5q14.3 in 15 subjects reported in this paper
Overall, dysmorphic features were not predominant, but some patients exhibited subtle hypertelorism, a “cupid-bow” shape to the upper lip, and hypotonic facies (Fig. 2a–g). The deletions in three subjects included RASA1, implicated in the “5q14.3 neurocutaneous syndrome” [2], but only one subject in our cohort (LR12-031) had characteristic capillary malformation of the skin (Fig. 2h). This subject also had atrophic skin adjacent to the suprasternal notch. All subjects had global developmental delay, and none had a phase of regression. All subjects had a normal head circumference.

Photographs of subjects. a IS09-024 at age 11 years. b LR11-305 at 12 months. c LR11-306 at 5 years, 3 months. d LR11-308 at 46 years. e LR11-309 at 17 months. f LR11-312 at 5 months. g LR11-325 at 6 months. Note characteristics of tenting of upper border of the mouth and hypotonic facies in several subjects. h Characteristic capillary malformation on the lower extremity of LR12-031
On brain imaging, there were no pathognomonic features, but several of the subjects had mild asymmetry of the lateral ventricles, increased periventricular white matter signal, dysgenesis of the corpus callosum, and mild cerebellar vermis hypoplasia (Fig. 3). Subject LR11-325, with a 6.0-Mb deletion of 5q14.3 extending into the critical region reported for periventricular nodular heterotopia [28], had a normal brain MRI.

Representative brain imaging findings. Subject LR11-047 has normal midline sagittal structures (a), and posterior periventricular white matter hyperintensities on axial T2 FLAIR (b). Subject LR11-307 has a dysmorphic corpus callosum and mild cerebellar vermis hypoplasia (c), with the cerebellar vermis disproportionately small compared to the cerebellar hemispheres on T2 coronal (d). Subject LR11-310 has normal midline sagittal structures (e) and mild thinning of the cortical white matter on T2 axial (f). Subject LR11-312 has frontal bossing and brachycephaly (g), as well as mild cortical atrophy and thinning of the white matter visible on T2 axial (h)
The most consistent neurologic features of MEF2C haploinsufficiency reported in the literature are (1) epilepsy, (2) abnormal movement patterns, and (3) a neurobehavioral “autism-plus” phenotype. We therefore focused our attention on these outcomes as the core features of MEF2C haploinsufficiency syndrome. Other clinical details of the 16 subjects are found in the Supplementary Clinical Data.
Epilepsy types and ages of onset
Ten of the 16 subjects (63 %) had epilepsy (defined as two or more unprovoked seizures), and six of these had onset of epilepsy in either infancy or soon after the first year of life. Representative EEG abnormalities are shown in Fig. 4. Two subjects had infantile spasms (classic epileptic spasms with hypsarrhythmia) alone, two subjects had myoclonic seizures with onset during infancy (myoclonic seizures, multifocal spikes on EEG, without hypsarrhythmia), and two subjects (LR11-307 and LR11-312) first presented with myoclonic seizures that then evolved into infantile spasms. The other four subjects with epilepsy had onset during childhood (defined as after 18 months of age), and these were intractable in all. Six of the 16 subjects had no diagnosis of epilepsy, including LR11-308 who at the age of 46 years had never had a seizure. Subjects LR11-387 and LR12-013 had multiple febrile seizures, but did not develop epilepsy. The four subjects with partial deletion of MEF2C (LR11-387, LR12-013, LR12-021, and LR12-022) did not have epilepsy, and this was statistically significant compared to the prevalence of epilepsy among our subjects with full deletion (p < 0.0005).

Representative EEG abnormalities in subjects with MEF2C haploinsufficiency showing spike-wave associated with epileptic spasm in subject IS09-024 (a) and multifocal epileptiform activity and poorly developed anterior-posterior gradient in subject LR11-310 (b)
When combined with the previously reported subjects with MEF2C haploinsufficiency, we confirm the frequent presence of infantile spasms and infant-onset myoclonic epilepsy (Table 2). The majority (54 % of all subjects) had an infant-onset epilepsy, either myoclonic seizures (33 %) or infantile spasms (21 %). Other subjects reported here and in the literature had childhood-onset epilepsy (24 %), with 12 % having intractable epilepsy and 12 % controlled by medication. A few patients (12 %) had febrile seizures, and across all patients reported to date, 23 % had no diagnosis of epilepsy.
Abnormal movement patterns
Nearly all (93 %) of the subjects in this report presented with hypotonia as a prominent feature during infancy, and then developed abnormal movements with paroxysms of excess motion best classified as hyperkinesis [22]. During infancy, this pattern may be subtle, but still discernible in some patients (Video 1). By later childhood, the hyperkinesis manifests as a series of rapid stereotypies that are prominent when the child is excited and may interrupt ongoing activity (Video 2). As nearly all of the children in this series had severe ID, it was difficult to assess whether the stereotypies could be voluntarily suppressed. However, the hyperkinetic movements varied in response to external stimuli. At times, the stereotypies were accompanied by dystonia or chorea. Parents and care providers commented on the omnipresence and often constant nature of the stereotypies. The hyperkinesis appears to moderate over time, as several care providers reported they lessened as the children aged. Video from 46-year-old subject LR11-308 was notable for hypokinetic spasticity with intermittent hand-wringing stereotypies (not shown).
Neurobehavioral and neurofunctional outcomes
We obtained detailed data on neurobehavioral and neurofunctional impairments in 14 of the 16 subjects with MEF2C haploinsufficiency, and the common features are shown in Table 3. Most subjects had significant abnormalities of gastrointestinal motility, including gastroesophageal reflux disease, dysphagia, and constipation. All subjects were averbal with normal hearing, indicating severe impairment of language development. Most parents reported their children were prone to inappropriate laughter. The exception was subject LR11-307 who was easily agitated and engaged in self-mutilating behaviors. Several subjects had a phase of early irritability during infancy, replaced by a generally happy demeanor. Most parents also reported their children had high pain tolerance, manifested as not crying during intramuscular immunizations or after injuries. Poor reciprocal behaviors were common, with lack of or avoidance of eye contact, poor visual tracking, and limited engagement with or recognition of other people. Six subjects had poor sleep initiation and/or maintenance. Breathing rhythm, satiety, and temperature regulation were reported as normal in all subjects, aside from subject LR11-307 who had persistent episodes of hyper/hypoventilation and LR12-013 who had frequent breath-holding behavior.
Analysis of published array data
In order to better understand the role of MEF2C in the developing forebrain, we examined the microarray data files from several publications that used expression arrays in mice designed to study genes putatively upstream of Mef2c expression [19, 20, 25]. We confirmed that Mef2c expression was diminished in the Dlx1/2 knockout mouse. Consistent with the report that MECP2 expression is diminished in MEF2C haploinsufficient patients [7], we found that Mecp2 expression is similarly reduced in the Dlx1/2 knockout mouse forebrain, suggesting a network relationship between Dlx1/2, Mef2c, and Mecp2 (Supplementary Tables 1 and 2). Unfortunately, no probes for Cdkl5 were present on the Affymetrix arrays used, so we could not evaluate Cdkl5 expression in the Dlx1/2 knockout mouse.
Mef2c expression and localization in mouse forebrain neuronal populations
Due to evidence that Mef2c is involved in the development of both dorsal glutamatergic neurons and ventral GABAergic interneurons, we assayed the neuronal cell-type specific Mef2c gene expression and staining pattern in the mouse forebrain. Mef2c expression was significant in migrating interneurons versus non-interneurons in the cortex at e14.5, when compared to GFP-positive control cells in the ganglionic eminence (GE), as summarized in Table 4. However, Mef2c staining was seen in both Dlx5/6(+) interneurons in the GE, as well as in neurons elsewhere in the cortex at e14.5 (Fig. 5). Staining for Mef2c at P14 revealed scattered neuronal labeling, but more prominent in the upper layers of the cortex. The immunohistochemical staining at both ages was in agreement with in situ data from published sources (http://developingmouse.brain-map.org/data/search/gene/index.html?term=Mef2c).

Mef2c is present in both developing excitatory and inhibitory neurons throughout development. At embryonic day 14 (e14.5), Mef2c-positive cells (red in A′, B′) are found in the developing cortical plate and basal ganglia. Dlx5/6-positive cells (developing interneurons; green in A″, B″) are present in the SVZ of the ganglionic eminence (GE), migrating into the cortex in the intermediate zone (IZ, thin bracket in B) and the marginal zone (MZ, labeled in A), as well as having entered the cortical plate (CP, thick bracket in B). There is co-localization between Mef2c(+) cells and Dlx5/6 cells, primarily in the MZ, but also in the CP and SVZ. Expansion of the boxed area in A (A′ and A″) is shown in B. There is more substantial colocalization in the forming basal ganglion (asterisk). This staining pattern for Mef2c is consistent with the Allen Brain Atlas at e14.5 (C) as is mostly found in the CP but also in the MZ (bracket and arrows in C). In the mature brain, Mef2c is located throughout the cortex both by in situ hybridization (brackets in D) and immunohistochemistry (E) and in interneurons in the hippocampus (arrows in D)
Discussion
MEF2C haploinsufficiency syndrome is an emerging neurodevelopmental disorder with only 27 previously published cases. One patient with deletion of a putative regulatory region upstream of MEF2C [29] and another patient with an unbalanced chromosomal translocation [30] have also been reported. We describe an additional 16 subjects with the goal of furthering the characterization of the neurologic phenotype seen in this disorder. The core phenotype includes consistent hyperkinesis, hypotonia, intellectual disability with deficits in verbal language, and alterations in GI motility, mood, pain tolerance, and social reciprocity. Surprisingly, though epilepsy is a major feature, it is not uniform but variable in both its severity and age of onset and not present in one-fifth of the patients.
From the combined phenotype data that includes our new patient series and those already published, a spectrum can be described ranging from those with severe epilepsy/consistent hyperkinesis to those with no epilepsy or milder epilepsy who nevertheless have consistent hyperkinesis. Only six out of 16 subjects in this report had infant-onset myoclonic seizures and/or electroclinically defined infantile spasms. An important observation from our data is that, despite the frequency of early-onset epilepsies, those children with later onset, milder seizures, or no epilepsy at all also had severe neurobehavioral and neurofunctional outcomes. While there was no obvious correlation between severity of the neurologic phenotype and deletion size, children in our cohort with partial deletion of MEF2C were significantly more likely not to have epilepsy. It is possible that effects on alternative splicing of the neuronal MEF2C isoform may affect the severity of epilepsy in subjects with MEF2C haploinsufficiency.
Hyperkinesis was seen consistently among the patients in this report, regardless of deletion size and whether epilepsy was present or not. The hyperkinesis observed in MEF2C haploinsufficiency patients is similar to the movement disorders described in children with a number of other developmental disorders due to a variety of genes. For example, children with ARX mutations often have a notable movement disorder [31, 32], and hyperkinesis and stereotypic movements have been documented in patients with both duplications and deletions of FOXG1 [33, 34]. Additionally, although patients with Rett syndrome have characteristic stereotyped movements [35, 36], the quality of these movements is distinct from those seen in individuals with MEF2C-related disorder. Together, we hypothesize that certain types of abnormal movements are a common feature among children with abnormalities of forebrain-expressed transcription factors, and the movement disorders seen in these patients may arise from deficits in a shared gene regulatory network.
MEF2C appears to be a marker neither for the development of dorsal-restricted nor ventral-restricted neuronal populations. Our meta-analysis of the gene expression data, as well as our immunostaining data across multiple stages of mouse forebrain development, suggest a more complex picture. While Mef2c expression is diminished in the context of Arx and Dlx1/2 deficiency, Mef2c staining is not restricted to ventral GABAergic interneuron populations. Mef2c expression during forebrain development therefore appears to begin in both dorsal primary neuroblasts and ventral GABAergic interneurons, but then during development is more prevalent among mature glutamatergic neurons. This is consistent with its known role as a marker of Tbr1-mediated cortical lamination [18]. This also confirms other forebrain gene expression studies where, although Mef2c was associated with two modules of interneuron development, the strength of these associations was insufficient to formally assign Mef2c to either of those modules [26]. The extent of interaction of Mef2c in the Dlx1/2–Arx pathway in GABAergic interneurons, the Tbr1 cortical neuron developmental pathway, Mecp2-associated pathways, and reported MEF2C interactions with CDKL5 and MECP2 are summarized in Fig. 6. Additionally, MEF2 interactions with the causative gene for Fragile X syndrome FMR1 appear to be important for post-developmental synaptic maturation [37]. The array and immunohistochemical data presented confirm the involvement of MEF2C in both dorsal and ventral neuronal development, and suggest a compound role for MEF2C in the generation of a complex neurodevelopmental disorder.

Summary of the published data showing gene regulatory relationships for Mef2c in the Dlx1/2–Arx pathway in GABAergic interneurons [19, 20] (a), the Tbr1 cortical neuron developmental pathway [18] (b), Mecp2-associated pathways [27] (c), and reported MEF2C interactions with CDKL5 and MECP2 [7], as well as interactions with FMR1 during synaptic development [37] (d)
We present a large series of MEF2C haploinsufficiency patients and provide detailed description of the neurologic phenotype. These data illustrate a recurring clinical profile of MEF2C haploinsufficiency distinct from other severe neurodevelopmental disorders such as Rett syndrome and FOXG1-related disorders. This profile specifically includes a consistent hyperkinetic movement disorder, intellectual disability with impairment of spoken language, reciprocal behavior, GI motility, mood, and pain tolerance. Breathing rhythm abnormalities were uncommon, and there were no obvious impairments of the autonomic nervous system. The types and severity of epilepsy are variable, and the absence of epilepsy is significantly correlated with partial deletion of MEF2C. Overall, these data suggest the phenotypic features of MEF2C haploinsufficiency may be a reflection of dysregulation of development of multiple neuronal cell populations at the transcriptional level.
References
Berland S, Houge G (2010) Late-onset gain of skills and peculiar jugular pit in an 11-year-old girl with 5q14.3 microdeletion including MEF2C. Clin Dysmorphol 19(4):222–224
Carr CW, Zimmerman HH, Martin CL, Vikkula M, Byrd AC, Abdul-Rahman OA (2011) 5q14.3 neurocutaneous syndrome: a novel continguous gene syndrome caused by simultaneous deletion of RASA1 and MEF2C. Am. J. Med. Genet. A 155A(7):1640–1645
Engels H, Wohlleber E, Zink A, Hoyer J, Ludwig KU, Brockschmidt FF et al (2009) A novel microdeletion syndrome involving 5q14.3-q15: clinical and molecular cytogenetic characterization of three patients. Eur J Hum Genet 17(12):1592–1599
Le Meur N, Holder-Espinasse M, Jaillard S, Goldenberg A, Joriot S, Amati-Bonneau P et al (2010) MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J Med Genet 47(1):22–29
Novara F, Beri S, Giorda R, Ortibus E, Nageshappa S, Darra F et al (2010) Refining the phenotype associated with MEF2C haploinsufficiency. Clin Genet 78(5):471–477
Nowakowska BA, Obersztyn E, Szymańska K, Bekiesińska-Figatowska M, Xia Z, Ricks CB et al (2010) Severe mental retardation, seizures, and hypotonia due to deletions of MEF2C. Am J Med Genet B Neuropsychiatr Genet 153B(5):1042–1051
Zweier M, Gregor A, Zweier C, Engels H, Sticht H, Wohlleber E et al (2010) Mutations in MEF2C from the 5q14.3q15 microdeletion syndrome region are a frequent cause of severe mental retardation and diminish MECP2 and CDKL5 expression. Hum Mutat 31(6):722–733
Mikhail FM, Lose EJ, Robin NH, Descartes MD, Rutledge KD, Rutledge SL et al (2011) Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am J Med Genet A 155A(10):2386–2396
Zweier M, Rauch A (2012) The MEF2C-related and 5q14.3q15 microdeletion syndrome. Mol Syndromol 2(3–5):164–70
Li Z, McKercher SR, Cui J, Nie Z, Soussou W, Roberts AJ et al (2008) Myocyte enhancer factor 2C as a neurogenic and antiapoptotic transcription factor in murine embryonic stem cells. J Neurosci 28(26):6557–6568
Potthoff MJ, Olson EN (2007) MEF2: a central regulator of diverse developmental programs. Development 134(23):4131–4140
Leifer D, Krainc D, Yu YT, McDermott J, Breitbart RE, Heng J et al (1993) MEF2C, a MADS/MEF2-family transcription factor expressed in a laminar distribution in cerebral cortex. Proc Natl Acad Sci U S A 90(4):1546–1550
Lyons GE, Micales BK, Schwarz J, Martin JF, Olson EN (1995) Expression of mef2 genes in the mouse central nervous system suggests a role in neuronal maturation. J Neurosci 15(8):5727–5738
Janson CG, Chen Y, Li Y, Leifer D (2001) Functional regulatory regions of human transcription factor MEF2C. Brain Res Mol Brain Res 97(1):70–82
Sekiyama Y, Suzuki H, Tsukahara T (2012) Functional gene expression analysis of tissue-specific isoforms of Mef2c. Cell Mol Neurobiol 32(1):129–139
Li H, Radford JC, Ragusa MJ, Shea KL, McKercher SR, Zaremba JD et al (2008) Transcription factor MEF2C influences neural stem/progenitor cell differentiation and maturation in vivo. Proc Natl Acad Sci U S A 105(27):9397–9402
Barbosa AC, Kim M-S, Ertunc M, Adachi M, Nelson ED, McAnally J et al (2008) MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc Natl Acad Sci U S A 105(27):9391–9396
Bedogni F, Hodge RD, Elsen GE, Nelson BR, Daza RAM, Beyer RP et al (2010) Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc Natl Acad Sci U S A 107(29):13129–13134
Fulp CT, Cho G, Marsh ED, Nasrallah IM, Labosky PA, Golden JA (2008) Identification of Arx transcriptional targets in the developing basal forebrain. Hum Mol Genet 17(23):3740–3760
Long JE, Cobos I, Potter GB, Rubenstein JLR (2009) Dlx1&2 and Mash1 transcription factors control MGE and CGE patterning and differentiation through parallel and overlapping pathways. Cereb Cortex 19(Suppl 1):i96–i106
Battini R, Sgandurra G, Petacchi E, Guzzetta A, Di Pietro R, Giannini MT et al (2008) Movement disorder-childhood rating scale: reliability and validity. Pediatr Neurol 39(4):259–265
Sanger TD, Chen D, Fehlings DL, Hallett M, Lang AE, Mink JW et al (2010) Definition and classification of hyperkinetic movements in childhood. Mov Disord 25(11):1538–1549
Ballif BC, Theisen A, McDonald-McGinn DM, Zackai EH, Hersh JH, Bejjani BA et al (2008) Identification of a previously unrecognized microdeletion syndrome of 16q11.2q12.2. Clin Genet 74(5):469–475
Marsh ED, Minarcik J, Campbell K, Brooks-Kayal AR, Golden JA (2008) FACS-array gene expression analysis during early development of mouse telencephalic interneurons. Dev Neurobiol 68(4):434–445
Batista-Brito R, Machold R, Klein C, Fishell G (2008) Gene expression in cortical interneuron precursors is prescient of their mature function. Cereb Cortex 18(10):2306–2317
Winden KD, Oldham MC, Mirnics K, Ebert PJ, Swan CH, Levitt P et al (2009) The organization of the transcriptional network in specific neuronal classes. Mol Syst Biol 5:291
Chahrour M, Jung SY, Shaw C, Zhou X, Wong STC, Qin J et al (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320(5880):1224–1229
Cardoso C, Boys A, Parrini E, Mignon-Ravix C, McMahon JM, Khantane S et al (2009) Periventricular heterotopia, mental retardation, and epilepsy associated with 5q14.3-q15 deletion. Neurology 72(9):784–792
Saitsu H, Igarashi N, Kato M, Okada I, Kosho T, Shimokawa O et al (2011) De novo 5q14.3 translocation 121.5-kb upstream of MEF2C in a patient with severe intellectual disability and early-onset epileptic encephalopathy. Am J Med Genet A 155(11):2879–84
Toral-López J, Buentello-Volante B, Balderas-Minor MM, Amezcua-Herrera C, Valdes-Miranda JM, González-Huerta LM et al (2012) An intellectually disabled patient with the 5q14.3q15 microdeletion syndrome associated with an apparently de novo t(2;5)(q13;q14). Am J Med Genet A 158A(4):942–946
Strømme P, Mangelsdorf ME, Scheffer IE, Gécz J (2002) Infantile spasms, dystonia, and other X-linked phenotypes caused by mutations in Aristaless related homeobox gene, ARX. Brain Dev 24(5):266–268
Guerrini R, Moro F, Kato M, Barkovich AJ, Shiihara T, McShane MA et al (2007) Expansion of the first PolyA tract of ARX causes infantile spasms and status dystonicus. Neurology 69(5):427–433
Brunetti-Pierri N, Paciorkowski AR, Ciccone R, Mina ED, Bonaglia MC, Borgatti R et al (2011) Duplications of FOXG1 in 14q12 are associated with developmental epilepsy, mental retardation, and severe speech impairment. Eur J Hum Genet 19(1):102–107
Kortüm F, Das S, Flindt M, Morris-Rosendahl DJ, Stefanova I, Goldstein A et al (2011) The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis. J Med Genet 48(6):396–406
Percy AK (2002) Rett syndrome. Current status and new vistas. Neurol Clin 20(4):1125–1141
Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N et al (2010) Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 68(6):944–950
Pfeiffer BE, Zang T, Wilkerson JR, Taniguchi M, Maksimova MA, Smith LN et al (2010) Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron 66(2):191–197
Tonk V, Kyhm JH, Gibson CE, Wilson GN (2011) Interstitial deletion 5q14.3q21.3 with MEF2C haploinsufficiency and mild phenotype: when more is less. Am J Med Genet A 155A(6):1437–1441
Bienvenu T, Diebold B, Chelly J, Isidor B (2012) Refining the phenotype associated with MEF2C point mutations. Neurogenetics. doi:10.1007/s10048-012-0344-7
Acknowledgments
We wish to thank the families of the subjects for sharing the details of their children’s condition with us. We recognize Natasha Vedage and Molly Bourke for their assistance with the immunohistochemistry, Erin Dodge for assistance with figure design, and Hailly Butler for assistance with subject consents.
Disclosures
WBD is funded by NINDS R01 NS058721; JAR and RAS are employees of Signature Genomic Laboratories, PerkinElmer; ANL is an employee of ARUP Laboratories; and RJV is an employee of Lineagen, Inc. The other authors have no disclosures.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOC 96 kb)
Video 1
Subject LR11-325 at 5 months, showing paroxysms of hyperkinetic movements of the arms and legs. These were not associated with epileptiform discharges on EEG. (MPG 7636 kb)
Subject IS09-024 at 12 years, showing stereotypic hyperkinetic movements of the distal upper extremities. (AVI 6156 kb)
Rights and permissions
About this article
Cite this article
Paciorkowski, A.R., Traylor, R.N., Rosenfeld, J.A. et al. MEF2C Haploinsufficiency features consistent hyperkinesis, variable epilepsy, and has a role in dorsal and ventral neuronal developmental pathways. Neurogenetics 14, 99–111 (2013). https://doi.org/10.1007/s10048-013-0356-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-013-0356-y