
Abstract
Junctophilin subtypes, designated as JPH1∼4, are protein components of junctional complexes and play essential roles in cellular Ca2+ signaling in excitable cells. Knockout mice lacking the cardiac-type Jph2 die of embryonic cardiac arrest, and the mutant cardiac myocytes exhibit impaired formation of peripheral couplings and arrhythmic Ca2+ signaling caused by functional uncoupling between dihydropyridine and ryanodine receptor channels. Based on these observations, we hypothesized that mutations of JPH2 could cause human genetic cardiac diseases. Among 195 Japanese patients (148 index cases and 47 affected family members) with hypertrophic cardiomyopathy (HCM), two heterozygous nonsynonymous nucleotide transitions, G505S and R436C, were newly found in JPH2. When Fisher’s exact test was used to compare index cases with HCM to unrelated Japanese healthy controls in the frequencies of mutant alleles, only the G505S mutation showed statistical significance (4/296 HCM patients and 0/472 control individuals, P=0.022). This result was still significant after Bonferroni’s correction for multiple comparisons (P=0.044). To the best of our knowledge, this is the first report on JPH2 mutation associated with HCM.
Similar content being viewed by others

Introduction
Functional communications between cell-surface and intracellular channels are essential features of excitable cells (Berridge 2002). During the contraction of striated muscle cells, the activation of cell-surface dihydropyridine receptor channels (DHPRs) opens ryanodine receptors (RyRs) and triggers Ca2+ release from the sarcoplasmic reticulum (SR) employing the “Ca2+-induced Ca2+ release (CICR)” or “voltage-induced Ca2+ release” mechanism (Meissner 1994). The functional couplings between the channels take place in junctional membrane complexes (JMCs), which are designated as the “triad junction” in skeletal muscle, “diad” in cardiac muscle, “peripheral coupling” in immature muscle and “subsurface cisterna” in neurons (Flucher 1992; Franzini-Armstrong and Protasi 1997). Recent studies have indicated that the junctophilin subtypes, JPH1 through 4, play essential roles in the formation of JMC in muscle cells (Takeshima et al. 2000; Komazaki et al. 2003; Nishi et al. 2003). In Jph1-knockout mice with perinatal lethality, mutant skeletal muscle shows a deficiency in triad junctions and insufficient contraction likely caused by impaired communication between DHPRs and RyRs (Ito et al. 2001). In Jph2-knockout embryos showing cardiac arrest, mutant cardiac myocytes bear a deficiency in peripheral couplings and arrhythmic Ca2+ signaling probably caused by functional uncoupling between DHPRs and RyRs (Takeshima et al. 2000; Uehara et al. 2002). In the brain, both Jph3 and Jph4 are expressed at similar discrete neuronal sites and may collaboratively contribute to JMC formation (Nishi et al. 2002, 2003). Knockout mice lacking both Jph3 and Jph4 show lethality during weaning stages under normal housing conditions (Moriguchi et al. 2006). Mutant hippocampal and cerebellar neurons from the double-knockout mice lack the after-hyperpolarization phase in action potential configuration, and Ca2+-mediated communication between cell-surface Ca2+ channels, RyRs and small-conductance Ca2+-activated K+ channels is disconnected likely due to JMC disassembly (Moriguchi et al. 2006; Kakizawa et al. 2007).
Mutations of JPHs may also cause human genetic diseases. Indeed, the insertions of triplet repeats in the JPH3 gene locus result in a human genetic disease called HDL2 revealing similar symptoms to Huntington’s disease (Holmes et al. 2001). On the other hand, mutations of cardiac Ca2+ signaling proteins are often responsible for cardiomyopathies. For example, arrhythmogenic right ventricular cardiomyopathy is associated with mutations of RYR2 encoding the cardiac ryanodine receptor (Tiso et al. 2001). Mutations of phospholamban are associated with hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) (Haghighi et al. 2003; Minamisawa et al. 2003; Schmitt et al. 2003). HCM is a primary myocardial disorder that results in hypertrophy of the ventricle wall histologically characterized by cardiac myocyte disarray and fibrosis (Elliott and McKenna 2004). The symptoms of HCM vary among patients from mild cases to critical conditions, including heart failure and sudden death (Maron 2002). DCM is characterized by dilation of the left ventricle and systolic dysfunction (Richardson et al. 1996). HCM and DCM are inherited in an autosomal dominant fashion in most cases (Taylor et al. 2004). On the basis of the observation that JPH2 is essential for murine cardiac function, we attempted to detect JPH2 mutations associated with cardiomyopathies. This paper is, to our best knowledge, the first report of a JPH2 mutation associated with HCM.
Materials and methods
Subjects
The study population consisted of 195 patients with HCM (148 index cases and 47 affected family members), including ten transition forms from HCM to DCM, 32 patients with DCM (29 index cases and three affected family members), and eight patients with restrictive cardiomyopathy (RCM) (seven index cases and one affected family member). Unaffected family members of the index cases who carry a JPH2 gene mutation were also examined for the mutation and clinical status. Control subjects were 236 unrelated healthy Japanese individuals randomly selected. All patients gave informed consent to the clinical and genetic studies. The internal ethics committee of Tokyo Women’s Medical University approved this study. The patients were evaluated by a detailed history, physical examination, electrocardiography (ECG) and echocardiography, which allowed the diagnosis of HCM to be made in those clinically affected. Individuals with another intrinsic cardiac or systemic disease capable of producing myocardial abnormalities were excluded.
Mutation screenings
Mutation screening was performed on the human JPH2 gene consisting of five coding exons, as shown in Fig. 1. Genomic DNA was extracted from peripheral blood lymphocytes of the patients as described previously (Yoshida et al. 1986). PCR was carried out by using 2× GC Buffer I (TAKARA BIO INC, Otsu, Japan) and AmpliTaq DNA polymerase (Applied Biosystems, Foster City, CA, USA). PCR primers used are shown as follows: 1F, AGGGCACTGGAGGAGTGTTG; 1R, ACCCTCCACCAGGGTTTCTC; 2-1F, CATAGCACTGTGCACCCTGA; 2-1R, GTTCTTCCACTCGCCCATGT; 2-2F, TAAGAGCGACCTCAGCTCGG; 2-2R, TGGAGACAGGGCCTCCTAGG; 3F, TTCATTGACTGCCTGCGTGA; 3R, CTCACGGGCATCCAGGAAAC; 4-1F, AGTGGCTGGCCCAGGATCAC, 4-1R, TCGGGCTGGTCCTCAAAGGG; 4-2F, GGGCAGCCGGTCAGTCACTC; 5F, GAACAAAGCCGGCAATGCTC; 5R, CTAGGTCTTGGCTTCTGCAGG. Amplified products were purified using a MultiScreen PCR plate (Millipore, Billerica, MA, USA) and directly sequenced using the ABI-PRISM Bigdye-terminator cycle sequencing reaction kit and ABI 3130xl genetic analyzer (Applied Biosystems). Proband patients bearing the JPH2 G505S mutation were additionally examined for HCM mutations reported so far, i.e., the coding regions of cardiac beta-myosin heavy chain (MYH7), cardiac myosin-binding protein C, cardiac troponin T, cardiac troponin I, alpha-tropomyosin, myosin regulatory light chain, myosin essential light chain, cardiac alpha-actin, caveolin-3, titin-cap, all exons and a promoter region of PLN, the 14th and 45th exons (in N2-A and N2-B variants, respectively) of titin, the 20th exon of alpha-myosin heavy chain, the third exon of muscle LIM protein, and the third exon of troponin C. Primer sequences and detailed PCR conditions for these additional analyses are available upon request. Reference sequences and SNP information were obtained from the National Center for Biotechnology Information (NCBI http://www.ncbinlm.nih.gov/Sequin/).

The schematic structure of JPH2. The JPH2 gene contains five protein-coding exons (E1∼5) indicated by boxes, and the coding and untranslated regions are shown by filled and open boxes, respectively. Primers for PCR screening in this study are illustrated by arrows. JPH2 protein is composed of six predicted domains, which are the MORN motif region I (MORN-1), joining region (Joining), MORN motif region II (MORN-2), putative α-helical region (Helix), divergent region (Divergent) and membrane-spanning region (MS). The nonsynonymous heterozygous nucleotide transitions found in this study are indicated by asterisks
Statistical analyses
Fisher’s exact test was performed using the R environment to determine P-values for the association between HCM and JPH2 mutations using contingency tables containing allele frequencies. The conservative Bonferroni’s correction for multiple comparisons was used for adjustment in multiple testing, in which the P-value obtained was multiplied by the number of tests performed. A P-value below 0.05 was considered as significant.
Peptide conformation analysis
Circular dichroic (CD) spectra were recorded on a Jasco J-720 spectropolarimeter using a 0.4-mm path-length quartz cuvette at 25°C. Stock solutions of synthetic peptides (Hokkaido System Science Co., Sapporo, Japan) were adjusted to 1 mM in phosphate buffered saline (PBS) and further diluted with PBS or 2,2,2-trifluoroethanol (TFE) to 50 mM for the analysis. The spectrum was obtained by averaging six successive individual scans over the 195 to 250-nm wavelength range and the buffer solvent backgrounds were subtracted.
Results and discussion
A case-control study was performed to determine the association between JPH2 and cardiomyopathies by analyzing sequence alterations of protein-coding exons including adjacent proximal intron regions in genomic DNAs (Fig. 1) extracted from 195 patients with HCM (148 index cases and 47 affected family members), 32 patients with DCM (29 index cases and three affected family members) and eight patients with RCM (seven index cases and one affected family member). As a result, we found two nonsynonymous heterozygous single nucleotide transitions, G1513A (G505S) and C1306T (R436C), in JPH2 in the patients with HCM. On the other hand, we could not find any sequence alternations in the patients with DCM or RCM. Portions of these transitions were subsequently analyzed in 236 unrelated healthy Japanese individuals. The former transition was detected in four unrelated patients (4/296 alleles), but was not detected in the healthy control subjects (0/472 alleles) (Fig. 2a). The frequency of this transition was statistically higher in the patients with HCM (index cases) than the controls (P=0.022, Fisher’s exact test). The latter transition was found in two unrelated patients (2/296 alleles) as well as in the healthy controls (2/472 alleles), and there was no significant difference between the patients and controls in the frequencies of the mutant alleles (P=0.500, Fisher’s exact test). Since we performed statistical tests for two different missense polymorphisms, the correction for the multiple comparisons should be applied. Even through the conservative Bonferroni’s correction for multiple comparisons, the P-value of 0.022 remains statistically significant since 0.044, which is lower than 0.05, is obtained when the P-value is multiplied by the number of tests performed. Therefore, the G505S substitution is closely associated with HCM, while the R436C substitution seems to be a polymorphism that is not associated with the disease.

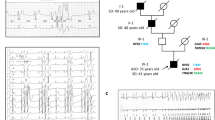
a DNA sequence analysis of JPH2 in a healthy control subject and the proband (II: 1) in family A. A heterozygous nonsynonymous nucleotide transition, G1513A (G505S), is shown in both the forward and reverse orientations. b Pedigree drawings of families A and B showing genotypes and phenotypes. Clinical state was determined independently of the genetic information. Arrows indicate probands in the families. The open boxes indicate men without HCM. The closed box indicates a man with HCM. The closed circles indicate women with HCM. The gray circle indicates a woman without a determined clinical condition. The dotted circle indicates a woman with other cardiac defects (see text). Plus and minus signs exhibit individuals with or without the G505S mutation of JPH2, respectively. The individual with a slash died. The asterisks show individuals with the A26V mutation of MYH7. The sharp shows individuals with the F513C mutation of MYH7. Age at diagnosis or sudden death is also noted. y and d represent years and day, respectively. SD sudden death
Of the four index patients with G505S, two had family members available for the genetic study (Fig. 2b). The proband in family A (II:1) was male and was diagnosed with HCM at 14 years of age. Electrocardiography (ECG) showed left ventricular hypertrophy (LVH), deep Q wave, and apparent ST-T changes. Myocardial imaging showed anterolateral hypertrophy of the free left ventricular wall. His mother (I:2) was also diagnosed with HCM at 40 years of age and carried the same genotype mutation. The ECG showed LVH, and echocardiography showed sigmoid septum and hyperdynamic LV motion. His healthy father (I:1) did not have the mutation allele. His younger sister (II:2) died suddenly of unknown causes at 3 years of age so she could not be examined in this study. The proband from family B (I:2) was female and she was diagnosed as having HCM at 33 years of age. She had a family history of HCM involving her grandfather, father, and the father’s siblings who were not available for genetic analysis. Echocardiography showed asymmetric hypertrophy of the interventricular septum. Her husband (I:1) had no sign of cardiac disease. An echocardiogram of her daughter (II:1) showed muscular ventricular septal defect and small atrial septal defect but no sign of HCM at 4 years of age. The defects caused little hemodynamic pathology. Her son was available for the mutation analysis and we found the same mutation, G505S, in the son. Echocardiography performed on the day after birth found no HCM sign in the son, although he may have a risk of HCM. One of the other two patients was male and was diagnosed as having HCM at 7 months of age. ECG showed LVH and echocardiography showed hypertrophy of the interventricular septum and posterior wall of left ventricle (both 15–16 mm), and left ventricular outflow tract obstruction. The other patient was also male and was diagnosed with HCM at 35 years of age. ECG showed negative T wave, abnormal Q wave, and nonsustained ventricular tachycardia. Echocardiography showed intraventricular septum hypertrophy of the left ventricle and paradoxical flow. Magnetic resonance imaging of the heart showed an apical left ventricular aneurysm. Histology of a myocardial biopsy of the right ventricle exhibited hypertrophy of the myocytes, disarray and myocardial fibrosis, which was consistent with a diagnosis of HCM (Fig. 3).

Histology of a biopsy specimen of myocardium from the right ventricle. a Masson’s trichrome was used to stain fibrosis blue (×10 in original magnification). b Myocyte hypertrophy and disarray are evident in hematoxylin and eosin-stained sections (×20 in original magnification)
Mutations of other predisposed genes for HCM might also be associated in these patients. In these patients, we further analyzed 15 genes known for association with HCM, including cardiac beta-myosin heavy chain (MYH7), cardiac myosin-binding protein C, and cardiac troponin T. We found heterozygous mutations of A26V and F513C of MYH7, which are known to be associated with HCM (Anan et al. 1994; Song et al. 2005), in the proband in family B (I:2). Her son (II:2) also carried the A26V mutation. These results suggested that both the G505S mutation of JPH2 and the mutations of MYH7 could be associated with the pathogenesis of HCM in the proband from family B.
JPH subtypes share characteristic structural features deduced from their hydropathicity profiles and predicted secondary structures. Based on the structural features, local sequence homology and genomic organization, six domain structures (Fig. 1) have been predicted in the JPH molecule (Nishi et al. 2000). Since the G505S mutation is located in the divergent region, in which no sequence homology is observed, and the numbers of amino acid residues are highly variable among JPH subtypes, it is unlikely that the G505S mutation affects the fundamental JPH functions including interaction with the plasma membrane and insertion into the SR membrane. A computer survey of the databases did not indicate that the G505S mutation would generate or break consensus motif sequences, such as phosphorylation and lipid-modification sites. Therefore, the G505S mutation could cause a structural defect indirectly influencing a certain physiological function of JPH.
To deduce the secondary structure of the region encompassing the G505S mutation in JPH2, wild-type and mutant peptides composed of 37 residues were synthesized and analyzed using CD spectroscopy (Fig. 4). The wild-type peptide showed a typical profile for random coil structure in PBS, i.e., no significantly positive peaks at 218 nm for β-strands or at 209 and 222 nm for α-helixes were detected. The organic solvent TFE is well known for its ability to induce secondary structures in peptides and proteins (Nelson and Kallenbach 1989; Sönnichsen et al. 1992). Although the precise mechanism of such conformation induction remains unclear, it is generally agreed that structure formation occurs due to enhanced intramolecular hydrogen-bonding capabilities in TFE-rich environments. However, α-helix- and β-strand-propensity inherent in the primary sequence is absolutely necessary for development of the secondary structure in the host peptide. The wild-type peptide in 80% TFE showed a broad and weak inflection at 210–230 nm and an attenuated 195-nm peak for random structures, suggesting that TFE promotes secondary structure formation. However, the TFE effect is remarkably weak and rather atypical. Moreover, no significant differences in the CD spectrum were detected between the mutant and wild-type peptides, suggesting that the structural effect of G505S mutation is minimal. Since the region surrounding G505S is enriched in proline residue (8/37 residues), this region may hardly fold secondary structures. JPH2 supports functional communication between DHPRs and RyRs and thus essentially contributes to Ca2+ signaling during cardiac contraction (Takeshima et al. 2000). Although our present results do not indicate that the G505S mutation results in the obvious structural abnormality of JPH2, it is still possible that the G505S mutation may produce a mild conformational change, weaken the efficiency of excitation-contraction, and further induce compensatory hypertrophy associated with altered metabolism in cardiomyocytes (Ashrafian et al. 2003). However, it remains to be investigated in future studies how the mutation alters JPH2 functions.

CD spectra of 37-mer JPH2 peptides with or without the G505S mutation in the center. The spectra were measured in PBS with and without 80% TFE. No significant difference was observed between the spectra of the wild-type and mutant peptides
References
Anan R, Greve G, Thierfelder L, Watkins H, McKenna WJ, Solomon S, Vecchio C, Shono H, Nakao S, Tanaka H, Mares A Jr, Towbin JA, Spirito P, Roberts R, Seidman JG, Seidman CE (1994) Prognostic implications of novel beta cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J Clin Invest 93:280–285
Ashrafian H, Redwood C, Blair E, Watkins H (2003) Hypertrophic cardiomyopathy: a paradigm for myocardial energy depletion. Trends Genet 19:263–268
Berridge MJ (2002) The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32:235–249
Elliott P, McKenna WJ (2004) Hypertrophic cardiomyopathy. Lancet 363:1881–1891
Flucher BE (1992) Structural analysis of muscle development: transverse tubules, sarcoplasmic reticulum, and the triad. Dev Biol 154:245–260
Franzini-Armstrong C, Protasi F (1997) Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol Rev 77:699–729
Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulous S, Liggett SB, Dorn GW, MacLennan DH, Kremastinos DT, Kranias EG (2003) Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 111:869–876
Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, Ross CA, Margolis RL (2001) A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 29:377–378
Ito K, Komazaki S, Sakamoto K, Yoshida M, Nishi M, Kitamura M, Takeshima H (2001) Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol 154:1059–1067
Kakizawa S, Kishimoto Y, Hashimoto K, Miyazaki T, Furutani K, Shimizu H, Fukaya M, Nishi M, Sakagami H, Ikeda A, Kondo H, Kano M, Watanabe M, Iino M, Takeshima H (2007) Junctophilin-mediated channel crosstalk essential for cerebellar synaptic plasticity. EMBO J 26:1924–1933
Komazaki S, Nishi M, Takeshima H (2003) Abnormal junctional membrane structures in cardiac myocytes expressing ectopic junctophilin type 1. FEBS Lett 542:69–73
Maron BJ (2002) Hypertrophic cardiomyopathy: a systematic review. JAMA 287:1308–1320
Meissner G (1994) Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu Rev Physiol 56:485–508
Minamisawa S, Sato Y, Tatsuguchi Y, Fujino T, Imamura S, Uetsuka Y, Nakazawa M, Matsuoka R (2003) Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy. Biochem Biophys Res Commun 304:1–4
Moriguchi S, Nishi M, Komazaki S, Sakagami H, Miyazaki T, Masumiya H, Saito S, Watanabe M, Kondo H, Yawo H, Fukunaga K, Takeshima H (2006) Functional uncoupling between Ca2+ release and afterhyperpolarization in mutant hippocampal neurons lacking junctophilins. Proc Natl Acad Sci USA 103:10811–10816
Nishi M, Mizushima A, Nakagawara K, Takeshima H (2000) Characterization of human junctophilin subtype genes. Biochem Biophys Res Commun 273:920–927
Nishi M, Hashimoto K, Kuriyama K, Komazaki S, Kano M, Shibata S, Takeshima H (2002) Motor discoordination in mutant mice lacking junctophilin type 3. Biochem Biophys Res Commun 292:318–324
Nishi M, Sakagami H, Komazaki S, Kondo H, Takeshima H (2003) Coexpression of junctophilin type 3 and type 4 in brain. Mol Brain Res 118:102–110
Nelson JW, Kallenbach NR (1989) Persistence of the alpha-helix stop signal in the S-peptide in trifluoroethanol solutions. Biochemistry 28:5256–5261
Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O’Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P (1996) Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 93:841–842
Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman EC (2003) Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299:1410–1413
Song L, Zou Y, Wang J, Wang Z, Zhen Y, Lou K, Zhang Q, Wang X, Wang H, Li J, Hui R (2005) Mutations profile in Chinese patients with hypertrophic cardiomyopathy. Clin Chim Acta 351:209–216
Sönnichsen FD, Van Eyk JE, Hodges RS, Sykes BD (1992) Effect of trifluoroethanol on protein secondary structure: an NMR and CD study using a synthetic actin peptide. Biochemistry 31:8790–8798
Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K (2000) Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell 6:11–22
Taylor MR, Carniel E, Mestroni L (2004) Familial hypertrophic cardiomyopathy: clinical features, molecular genetics and molecular genetic testing. Expert Rev Mol Diagn 4:99–113
Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A (2001) Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 10:189–194
Uehara A, Yasukochi M, Imanaga I, Nishi M, Takeshima H (2002) Store-operated Ca2+ entry uncoupled with ryanodine receptor and junctional membrane complex in heart muscle cells. Cell Calcium 31:89–96
Yoshida MC, Satoh H, Sasaki M, Semba K, Yamamoto T, Toyoshima K (1986) Regional location of a novel yes-related proto-oncogene, syn, on human chromosome 6 at band q21. Jpn J Cancer Res 77:1059–1061
Acknowledgments
We are grateful to Dr. Bernardo Nadal-Ginard for his valuable comments and Dr. Katsumi Matsuzaki for his kind support on CD measurements. We thank Dr. Kazuo Momma for providing clinical information of the patients. We also thank Drs. Tsutomu Nishizawa, Shin-ichiro Imamura, Shoichi Arai, Yoshiyuki Furutani, Ms. Michiko Furutani, and Mr. Hiroaki Nagao for their excellent technical assistance, and Ms. Barbara Levene for editing the manuscript. This work was supported by the Program for Promoting the Establishment of Strategic Research Centers, Special Coordination Funds for Promoting Science and Technology, Ministry of Education, Culture, Sports, Science and Technology (Japan), a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology (Japan), the Naito Foundation, the Sumitomo Foundation, and the Uehara Memorial Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Matsushita, Y., Furukawa, T., Kasanuki, H. et al. Mutation of junctophilin type 2 associated with hypertrophic cardiomyopathy. J Hum Genet 52, 543–548 (2007). https://doi.org/10.1007/s10038-007-0149-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-007-0149-y
Keywords
This article is cited by
-
T-tubule remodeling in human hypertrophic cardiomyopathy
Journal of Muscle Research and Cell Motility (2021)
-
Sacral agenesis: a pilot whole exome sequencing and copy number study
BMC Medical Genetics (2016)
-
Investigation of Pathogenic Genes in Chinese sporadic Hypertrophic Cardiomyopathy Patients by Whole Exome Sequencing
Scientific Reports (2015)
-
Mutations in the cardiac troponin T gene show various prognoses in Japanese patients with hypertrophic cardiomyopathy
Heart and Vessels (2013)
-
Emerging role of junctophilin-2 as a regulator of calcium handling in the heart
Acta Pharmacologica Sinica (2010)
