
Abstract
We report a patient with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome with a novel splicing mutation of the FOXP3 gene. The patient is a boy, born at 39 + 2 weeks gestation with a birth weight of 3,280 g. The family history was unremarkable. He was well until 11 months of age, when he was diagnosed with type 1 diabetes mellitus. The level of urine C-peptide was 0.58 μg/day (normal range, 44–116 μg/day). Glutamic acid decarboxylase autoantibody was not detected, but a high level of anti-insulin antibody (50 IU/mL; normal range, <5 IU/mL) was noted. This patient presented with unusual clinical features, including pure red cell aplasia, membranous glomerulopathy, and posterior reversible encephalopathy syndrome after a vaccination against influenza A H1N1 virus. The diagnosis of IPEX was made when the patient was 11 years old, which is quite late compared with typical cases. Conclusion: Although IPEX syndrome is usually a disease of infancy, it should not be ruled out solely on the basis of age. IPEX presentation is so variable that it should be suspected in a male child with one or more autoimmune disorders and severe infections.
Similar content being viewed by others


Introduction
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX; OMIM #304930) syndrome is a rare X-linked recessive disorder, usually manifesting as a multisystemic autoimmune disorder during infancy [9, 25]. The most common clinical features are severe secretory diarrhea, type 1 diabetes mellitus (T1DM), and eczema; less common symptoms include thyroiditis, hemolytic anemia, thrombocytopenia, autoimmune hepatitis, and recurrent infections [9, 25]. Abnormal development and/or function of CD4+CD25+ regulatory T cells caused by a mutation in the FOXP3 gene located on the short arm of the X chromosome has been shown to lie at the center of the pathogenesis [22]. Most patients have been reported to die within the first 2 years of life due to severe infection and/or failure to thrive, but some mid-to-long-term survivors have also been described [14, 20, 25].
Case report
This boy was born at 39 + 2 weeks gestation with a birth weight of 3,280 g. The family history was unremarkable. He was well until 11 months of age, when he was diagnosed with T1DM. The level of urine C-peptide was 0.58 μg/day (normal range, 44–116 μg/day). Glutamic acid decarboxylase autoantibody was not detected, but high level of anti-insulin antibody was detected (50 IU/mL; normal range, <5 IU/mL).
At 39 months, he was admitted to our institute for sudden pallor and decreased activity. There were no other signs of infection or bleeding. His plasma hemoglobin concentration was 2.3 mg/dL (normal range, 10.5–14.0 g/dL) with normal white blood cells and platelet counts. The reticulocyte production index was 0.42%. Serum concentrations of aspartate aminotransferase, alanine aminotransferase, and total bilirubin were 41 U/L (normal range, 15–55 U/L), 13 U/L (normal range, 5–45 U/L), and 0.4 mg/dL (normal range, <1.0 mg/dL), respectively. The serum concentration of lactate dehydrogenase (LDH) was 1,016 IU/L (normal range, 150–300 U/L) and that of haptoglobin was 98.4 mg/dL (normal range, 40–180 mg/dL). Direct and indirect Coombs test results were 3+ and 1+, respectively. A peripheral blood smear showed severe normocytic normochromic anemia without any evidence of hemolysis. One week after receiving a packed RBC transfusion, the patient's hemoglobin level was elevated to 11.9 g/dL.
A routine urinalysis revealed totally unexpected finding: albumin was 3+ and many RBCs were evident in a high-power-field microscopic examination. Twenty-four-hour urinary protein was 8,178 mg/day. Serum creatinine concentration was 0.4 mg/dL (normal range, 0.3–0.7 mg/dL), and the concentration of serum albumin was 2.5 g/dL (normal range, 3.7–5.6 g/dL). A renal biopsy confirmed the diagnosis of membranous glomerulonephritis (MGN). Serologic markers for hepatitis B and C infections were all negative.
Six weeks later, his hemoglobin dropped to 4.6 g/dL with reticulocytopenia. Direct/indirect Coombs test results were 1+ and negative, respectively; again, peripheral blood cell morphology showed no hemolytic features. Serum LDH and haptoglobin were 581 IU/L and 138 mg/dL, respectively. Bone marrow cellularity was normal for age but no erythroid precursors were found. Parvovirus was not detected by polymerase chain reactions of an aspiration sample of the bone marrow. Because the patient's anemia with reticulocytopenia had persisted for 4 weeks, he was diagnosed with pure red cell aplasia (PRCA) and started on steroid treatment with 2 mg/kg/day of prednisolone (PD). During the next 4 weeks, his hemoglobin level began to increase but was dependent on steroids. Over the next year, he was slowly tapered off with steroids and has since been maintained on a very low dose of PD (≤0.05 mg/kg/day). Hemoglobin levels have remained stable at 10.0–11.5 g/dL without transfusion. Although the patient's proteinuria had persisted, we did not give him other treatment for MGN. Besides proteinuria, there were no other signs of kidney dysfunction and PD had been administrated to the patient for the treatment of PRCA. We also followed up to check the persisting proteinuria with normal renal function, while PD was administered chronically for the treatment of PRCA.
At 7 years, the patient developed severe lobar pneumonia of probable bacterial origin, though the causative organism was not identified. Ampicillin–clavulanate was administered, and the patient was fully recovered after 2 weeks. His serum creatinine level was elevated to 1.9 mg/dL during antibiotic treatment, but returned to normal (0.4–0.5 mg/dL) after discontinuing antibiotics. A follow-up renal biopsy showed aggravated MGN compared with the previous biopsy.
At 11 years, the patient was hospitalized with meningitis. Bacterial meningitis was suspected based on CSF test results (WBC, 2,000/μL, of which 91% were neutrophils), although attempts to identify the causative organism failed. The patient was treated empirically with antibiotics, vancomycin, and ceftriaxone. His serum creatinine concentration was 1.8 mg/dL at admission, but rose to 9.0 mg/dL 2 weeks after the start of vancomycin administration. A renal biopsy revealed MGN with extensive interstitial inflammation, fibrosis, and chronic tubular atrophy compared with that previously observed. The vancomycin dose was adjusted to the level of serum creatinine, and the patient recovered from meningitis after a 3-week course of therapy. Serum creatinine levels decreased slowly after stopping the antibiotics and have since been maintained at 1.3–1.7 mg/dL.
The patient's clinical history, including early-onset diabetes, anemia (possibly related to autoimmune pathology), MGN, and severe infections, was suggestive of IPEX syndrome. A DNA analysis of his FOXP3 gene identified a novel mutation, c.201 + 1G>A [IVS1 + 1G>A], leading to a splicing error (Fig. 1). The same mutation in the FOXP3 gene was found in heterozygous form in his mother but was not detected in his younger brother. Notably, hypereosinophilia had been detected only when he had suffered from infection, without elevated level of immunoglobulin E.

Mutation analysis of the FOXP3 gene in our patient. Eleven exons and exon–intron boundaries were directly sequenced. The guanine residue at the splicing donor site of the first intron was replaced by an adenine, which may cause abnormal splicing
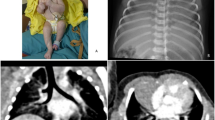
Four months after recovery from meningitis, and a week after receiving a vaccine against pandemic influenza A H1N1 virus, the patient developed posterior reversible encephalopathy syndrome (PRES) with sudden and severe hypertension (226/103 mmHg) (Fig. 2). When the patient was admitted, the serum concentration of creatinine was 1.2 mg/dL and that of blood urea nitrogen 16 mg/dL (normal range, 10–20 mg/dL). Although 24-hour urinary protein was 3,539.4 mg, serum albumin concentration was 3.2 g/dL and the patient showed no edema. On a renal Doppler ultrasonogram, cortical echogenicity was increased with loss of corticomedullary differentiation in both kidneys, but stenosis in either of the main renal arteries was not observed. Following antihypertensive therapy, his neurological findings returned to normal, and he was discharged without any symptoms after 2 weeks. His blood pressure has been maintained at <125/70 mmHg (90th percentile of boys of the same age) with enalapril (5 mg twice daily).

Magnetic resonance imaging findings. T2-weighted image (a) and fluid-attenuated inversion recovery (FLAIR) image (b) showing irregular high signal intensities on the white matters of bilateral posterior frontoparietal lobes and posterior corpus callosum, consistent with PRES
Discussion
In its most severe form, IPEX syndrome presents very early in life with multiple autoimmune disorders, classically T1DM and severe enteropathy [9, 25]. Additional features may include hemolytic anemia, thrombocytopenia, hypothyroidism, skin lesions, and recurrent infections [14, 25]. These disorders often appear sequentially and the affected organ spectrum can vary substantially from patient to patient [9]. Our patient was diagnosed with T1DM with anti-insulin antibody at 11 months, as a previously described IPEX patient [2]. He had loose bowel movements three to four times a day, but not watery diarrhea, which, if present, would have been very helpful for early diagnosis.
Several IPEX cases with renal involvement have been described, including proteinuria [1, 10, 13, 14, 16] and electrolyte imbalance due to tubular dysfunction [11]. Renal pathology in these patients was variable, including MGN [13], minimal change nephrotic syndrome [10], and/or focal tubular atrophy and tubulointerstitial nephritis [11, 13]. Renal function was normal in all of these patients, at least when the proteinuria began. Our patient showed heavy proteinuria with MGN pathology since infancy. A follow-up renal biopsy revealed interstitial inflammation and chronic tubular atrophy, although this finding might be resulted from the administration of a nephrotoxic drug, such as vancomycin.
Although most patients with IPEX syndrome who have anemia have autoimmune hemolytic anemia [14, 15, 17], our patient did not. Rather, both peripheral blood smears and laboratory findings revealed severe normocytic anemia with reticulocytopenia and few hemolytic features. Erythroid precursors were absent in otherwise normal bone marrow, and immunosuppressive therapy with a steroid improved his anemia. These features of the patient's anemia are compatible with those of PRCA, although the age at onset was very young [18, 26]. Immune-mediated erythropoietic failure as one of several pathogenetic mechanisms of PRCA is supported by clinical and laboratory evidence, which has shown that both antibody-mediated and cellular mechanisms are involved [26]. Because unregulated autoantibodies are closely related to the clinical presentation in IPEX syndrome, autoantibody-dependent PRCA may occur in patients with IPEX.
Our patient experienced PRES after vaccination against H1N1 influenza virus. Nearly all PRES cases have occurred in the setting of hypertension and/or use of immunosuppressive medications, especially cyclosporine [19]. Prior to the diagnosis of PRES, our patient had been normotensive and was not treated with cyclosporine. Although the patient had renal disease and persistent proteinuria, the reason for an abrupt onset of hypertension in such an individual with stable chronic renal disease is difficult to determine. However, the sudden development of hypertension occurred following vaccination. Abnormal reactions to vaccinations have been reported in patients with IPEX syndrome [14, 25]. It is likely that the vaccination triggered an acute dysregulated immune response and increasing immune complexes in this child leading to acute exacerbation of the underlying immune-mediated glomerulonephritis. In addition, his kidney function had deteriorated subsequent to the previous treatment of meningitis with vancomycin. It is reasonable to postulate that these factors, taken together, contributed to the development of sudden hypertension and PRES.
The causative gene of IPEX syndrome is FOXP3, which is located on chromosome region Xq11.23-Xq13.3 [5, 24]. The FOXP3 protein is a key regulatory factor in the development and function of CD4+CD25+ regulatory T cells [27]. More than 30 mutations in FOXP3 have been identified in patients with IPEX. Many of these are missense mutations within the forkhead (FKH) DNA-binding domain, but other mutations have been found throughout this gene [22, 27]. Mutations in the FKH domain interfere with nuclear import and DNA binding, both of which are critical for FOXP3 repressor activity, whereas mutations in the leucine zipper impair FOXP3 dimerization and DNA binding [22, 27]. Mutations outside FOXP3 coding regions, for example within an intron/exon splice junction or in the first polyadenylation signal of the gene, may interfere with normal gene expression and protein production [4, 6, 7, 9, 21, 23]. These latter mutations may also be associated with more attenuated phenotypes [9].
The FOXP3 mutation in our patient was located in the probable splice donor site of the first intron [IVS1+1G>A], a mutation not previously reported; this location may explain the relatively mild clinical presentation of our patient. However, there seems to be no clear-cut genotype–phenotype correlation in IPEX syndrome: some patients with a mutation within the FKH domain exhibit milder phenotypes [16], whereas other patients with a mutation outside the coding region have shown severe phenotypes, including early death [9, 23]. In addition, An et al. [1] described patients that shared the same FOXP3 mutation but exhibited different clinical features. Similarly, one patient reported by Gambineri et al. [9], whose mutation [IVS1+2T>G] was very close to that of our patient, showed quite different clinical features including severe diarrhea with intestinal villous atrophy, T1DM, and chronic eczema. Gambineri et al. [9] also reported that the correlation between FOXP3 expression and disease severity was inconsistent. In the study of An et al. [1], the frequency of CD4+CD25+ regulatory T cells did not differ between the IPEX patient and his normal twin brother. These observations suggest that a confirmative diagnosis of IPEX syndrome should be made through a mutation analysis of the FOXP3 gene [9]. Because we did not measure FOXP3 expression levels or CD4+CD25+ regulatory T cell counts, we cannot establish how our patient's FOXP3 mutation contributes to the development of his clinical features. It is unclear at present whether differences in clinical manifestations among various genotype–phenotype pairs are the result of chance, alternative genetic mechanisms, or the action of modifying genes, such as human leukocyte antigens (HLA). Environmental influences and variable management may also influence outcomes [25].
At present there are two main treatment modalities for IPEX syndrome: chronic immunosuppression and hematopoietic stem cell transplantation (HSCT) [25]. Chronic immunosuppression is partially effective in some patients but ineffective in others, varies by regimen, and increases the risks of severe and/or opportunistic infections. Generally, the prognosis of a patient with severe IPEX syndrome is very poor; in these patients, HSCT has been attempted, with some success [3, 12, 13, 15]. Interestingly, not only HSCT but the conditioning regimen itself has been shown to cause remission of nearly all clinical features, including diarrhea, diabetes, eczema, and proteinuria [3, 12, 13].
Our patient was 13 years old at the time of the submission of this manuscript and is in good clinical condition. He has not been hospitalized for the last 18 months and leads a nearly normal everyday life. He has been treated with insulin therapy since he was 11 months old, a very low dose of PD since 42 months old, and enalapril since 11 years old. Hemoglobin A1c has been maintained at <9%. However, his growth has been stunted and Cushingoid features have appeared within the last 2 years. His bone age has been delayed 3 years behind his chronological age. Heavy proteinuria without other nephrotic signs has persisted and serum creatinine has increased, although slowly. Because of this, the patient has been scheduled to receive HSCT from an 8/8 HLA-matched, unrelated donor.
In conclusion, although IPEX syndrome is usually a disease of infancy, it should not be ruled out solely on the basis of age. IPEX presentation is so variable that it should be suspected in a male child with one or more autoimmune disorders and severe infections [8]. The diagnosis would be confirmed by identifying a mutation in the FOXP3 gene.
References
An YF, Xu F, Wang M, Zhang ZY, Zhao XD (2011) Clinical and molecular characteristics of immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome in China. Scand J Immunol. doi:10.1111/j.1365-3083.2011.02574.x
Bacchetta R, Passerini L, Gambineri E, Dai M, Allan SE, Perroni L, Dagna-Bricarelli F, Sartirana C, Matthes-Martin S, Lawitschka A, Azzari C, Ziegler SF, Levings MK, Roncarolo MG (2006) Defective regulatory and effector T cell functions in patients with FOXP3 mutations. J Clin Invest 116:1713–1722
Baud O, Goulet O, Canioni D, Le Deist F, Radford I, Rieu D, Dupuis-Girod S, Cerf-Bensussan N, Cavazzana-Calvo M, Brousse N, Fischer A, Casanova JL (2001) Treatment of the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) by allogeneic bone marrow transplantation. N Engl J Med 344:1758–1762
Bennett CL, Brunkow ME, Ramsdell F, O’Briant KC, Zhu Q, Fuleihan RL, Shigeoka AO, Ochs HD, Chance PF (2001) A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA→AAUGAA) leads to the IPEX syndrome. Immunogenetics 53:435–439
Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD (2001) The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27:20–21
Costa-Carvalho BT, Moraes-Pinto MI, Almeida LC, Alves MT, Maia RP, Souza RL, Barreto M, Louren L, Vicente AM, Coutinho A, Carneiro-Sampaio M (2008) A remarkable depletion of both naive CD4+ and CD8+ with high proportion of memory T cells in an IPEX infant with a FOXP3 mutation in the forkhead domain. Scand J Immunol 68:85–91
De Benedetti F, Insalaco A, Diamanti A, Cortis E, Muratori F, Lamioni A, Carsetti R, Cusano R, De Vito R, Perroni L, Gambarara M, Castro M, Bottazzo GF, Ugazio AG (2006) Mechanistic associations of a mild phenotype of immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Clin Gastroenterol Hepatol 4:653–659
De Vries E, Driessen G (2011) Educational paper: primary immunodeficiencies in children: a diagnostic challenge. Eur J Pediatr 170:169–177
Gambineri E, Perroni L, Passerini L, Bianchi L, Doglioni C, Meschi F, Bonfanti R, Sznajer Y, Tommasini A, Lawitschka A, Junker A, Dunstheimer D, Heidemann PH, Cazzola G, Cipolli M, Friedrich W, Janic D, Azzi N, Richmond E, Vignola S, Barabino A, Chiumello G, Azzari C, Roncarolo MG, Bacchetta R (2008) Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol 122(1105–1112):e1101
Hashimura Y, Nozu K, Kanegane H, Miyawaki T, Hayakawa A, Yoshikawa N, Nakanishi K, Takemoto M, Iijima K, Matsuo M (2009) Minimal change nephrotic syndrome associated with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr Nephrol 24:1181–1186
Kobayashi I, Shiari R, Yamada M, Kawamura N, Okano M, Yara A, Iguchi A, Ishikawa N, Ariga T, Sakiyama Y, Ochs HD, Kobayashi K (2001) Novel mutations of FOXP3 in two Japanese patients with immune dysregulation, polyendocrinopathy, enteropathy, X linked syndrome (IPEX). J Med Genet 38:874–876
Mazzolari E, Forino C, Fontana M, D’Ippolito C, Lanfranchi A, Gambineri E, Ochs H, Badolato R, Notarangelo LD (2005) A new case of IPEX receiving bone marrow transplantation. Bone Marrow Transplant 35:1033–1034
Moudgil A, Perriello P, Loechelt B, Przygodzki R, Fitzerald W, Kamani N (2007) Immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: an unusual cause of proteinuria in infancy. Pediatr Nephrol 22:1799–1802
Powell BR, Buist NR, Stenzel P (1982) An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr 100:731–737
Rao A, Kamani N, Filipovich A, Lee SM, Davies SM, Dalal J, Shenoy S (2007) Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood 109:383–385
Rubio-Cabezas O, Minton JA, Caswell R, Shield JP, Deiss D, Sumnik Z, Cayssials A, Herr M, Loew A, Lewis V, Ellard S, Hattersley AT (2009) Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care 32:111–116
Satake N, Nakanishi M, Okano M, Tomizawa K, Ishizaka A, Kojima K, Onodera M, Ariga T, Satake A, Sakiyama Y (1993) A Japanese family of X-linked auto-immune enteropathy with haemolytic anaemia and polyendocrinopathy. Eur J Pediatr 152:313–315
Sawada K, Hirokawa M, Fujishima N (2009) Diagnosis and management of acquired pure red cell aplasia. Hematol Oncol Clin North Am 23:249–259
Stott VL, Hurrell MA, Anderson TJ (2005) Reversible posterior leukoencephalopathy syndrome: a misnomer reviewed. Intern Med J 35:83–90
Taddio A, Faleschini E, Valencic E, Granzotto M, Tommasini A, Lepore L, Andolina M, Barbi E, Ventura A (2007) Medium-term survival without haematopoietic stem cell transplantation in a case of IPEX: insights into nutritional and immunosuppressive therapy. Eur J Pediatr 166:1195–1197
Torgerson TR, Linane A, Moes N, Anover S, Vo M, Fi R-L, Hermine O, Vijay S, Gambineri E, Cerf-Bensussan N, Fischer A, Ochs HD, Goulet O, Ruemmele FM (2007) Severe food allergy as a variant of IPEX syndrome caused by a deletion in a noncoding region of the FOXP3 Gene. Gastroenterology 132:1705–1717
Van der Vliet HJ, Nieuwenhuis EE (2007) IPEX as a result of mutations in FOXP3. Clin Dev Immunol 2007:89017
Wang J, Li X, Jia Z, Tian Y, Yu J, Bao L, Wu Y, Ni B (2010) Reduced FOXP3 expression causes IPEX syndrome onset: an implication from an IPEX patient and his disease-free twin brother. Clin Immunol 137:178–180
Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME (2001) X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 27:18–20
Wildin RS, Smyk-Pearson S, Filipovich AH (2002) Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. J Med Genet 39:537–545
Young N (2006) Pure red cell aplasia. In: Lightman M, Beutler E, Kipps M, Seligsohn U, Kaushansky K, Prchal J (eds) Williams hematology, 7th edn. McGraw-Hill, New York, pp 437–447
Ziegler SF (2006) FOXP3: of mice and men. Annu Rev Immunol 24:209–226
Conflict of interests
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bae, K.W., Kim, B.E., Choi, JH. et al. A novel mutation and unusual clinical features in a patient with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Eur J Pediatr 170, 1611–1615 (2011). https://doi.org/10.1007/s00431-011-1588-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-011-1588-1