
Abstract
Transactive response DNA-binding protein 43 kDa (TDP-43) was identified as a major disease-associated component in the brain of patients with amyotrophic lateral sclerosis (ALS), as well as the largest subset of patients with frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U), which characteristically exhibits cytoplasmic inclusions that are positive for ubiquitin but negative for tau and α-synuclein. TDP-43 pathology occurs in distinct brain regions, involves disparate brain networks, and features accumulation of misfolded proteins in various cell types and in different neuroanatomical regions. The clinical phenotypes of ALS and FTLD-TDP (FTLD with abnormal intracellular accumulations of TDP-43) correlate with characteristic distribution patterns of the underlying pathology across specific brain regions with disease progression. Recent studies support the idea that pathological protein spreads from neuron to neuron via axonal transport in a hierarchical manner. However, little is known to date about the basis of the selective cellular and regional vulnerability, although the information would have important implications for the development of targeted and personalized therapies. Here, we aim to summarize recent advances in the neuropathology, genetics and animal models of TDP-43 proteinopathy, and their relationship to clinical phenotypes for the underlying selective neuronal and regional susceptibilities. Finally, we attempt to integrate these findings into the emerging picture of TDP-43 proteinopathy, and to highlight key issues for future therapy and research.
Similar content being viewed by others


Introduction
The pathology of proteinopathies appears in distinct brain regions, involves disparate brain networks, and features accumulation of misfolded proteins in various cell types and in different neuroanatomical regions. The clinical phenotypes of these diseases correlate with the characteristic distribution patterns of the underlying pathology across specific brain regions with disease progression. Individuals may exhibit rapid or slow neurodegeneration even within the same syndrome. However, little is known to date about the reasons for this, although an understanding of the basis of the selective cellular and regional vulnerability would be helpful for the development of targeted and personalized therapies. Notably, there is growing evidence that these proteinopathies are modulated by genetic risk factors, differences in the conformation of pathological proteins, and various environmental factors. These factors may play a role in differential cellular susceptibility or resilience, and hence result in phenotypic diversity.
Transactive response DNA-binding protein 43 kDa (TDP-43) was identified as a major disease-associated component in the brain of patients with amyotrophic lateral sclerosis (ALS) and the largest subset of patients with frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U) [6, 116]. FTLD-U has characteristic cytoplasmic inclusions, which are positive for ubiquitin but negative for tau and α-synuclein. The terminology FTLD-TDP has been recommended for the neuropathological subtype of FTLD characterized by abnormal intracellular accumulations of TDP-43 [44, 48, 99, 102, 114, 143].
In this review, we summarize recent advances in the neuropathology, genetics and animal models of TDP-43 proteinopathy, and discuss their relationship to clinical phenotypes, their place in the emerging picture of TDP-43 proteinopathy, and their relevance to the development of therapeutic interventions.
Cellular and molecular basis of TDP-43 aggregation
Structure, function and localization of TDP-43
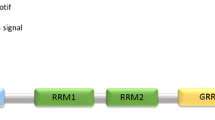
TDP-43 was originally discovered in a screen for protein factors that bind the long terminal repeat transactive response element of HIV-1 [119]. Several studies have revealed that TDP-43 can bind both DNA and RNA [31, 32, 94]. There is evidence that TDP-43 plays a role in transcription, alternative splicing and messenger RNA (mRNA) stability, and is involved in various cellular processes, including apoptosis, cell division and axonal transport [31, 94]. The capacity of TDP-43 to interact with RNA, including mRNA and pre-mRNA, is considered to underlie the role in exon splicing. TDP-43 is a heterogeneous nuclear ribonucleoprotein with two RNA recognition motifs (RRM-1 and 2) in the middle portion [93] and a glycine-rich domain and glutamine/asparagine (Q/N)-rich domain in the C-terminal region. Most mutations in TDP-43 are clustered in the C-terminal region [87], which is essential for some of the most well-characterized functions of TDP-43, including the protein’s role as a gene splicing factor [119] (Fig. 1).

Schematic diagram of TDP-43 functional regions involved in the pathogenesis and progression of TDP proteinopathy. TDP-43 in human brain is a heterogenous ribonucleoprotein with two RNA recognition motifs (RRM-1 and 2) in the middle portion and with a glycine-rich domain and Q/N-rich domain in the C-terminal region. Protein in abnormal conformations is accumulated in brains of patients in the form of amyloid-like fibril structures. The blue arrow indicates the protease-resistant fibril core. Mapping of three selected antibodies to the TDP-43 protein is shown. Antibodies in italics are widely used in TDP-43 immunohistochemical studies [62]. NLS nuclear localization signal, RRM RNA-recognition motif, Gly-rich glycine-rich domain
TDP-43 shuttles between the nucleus and cytoplasm under physiological conditions, and a nuclear localization signal enables the active import of the protein into the nucleus [93, 159]. The majority of TDP-43 resides in the nucleus, but up to 30% of TDP-43 resides in the cytoplasm [17]. Export is mainly driven by passive diffusion, and TDP-43 is predominantly localized in the nucleus in the healthy state [93]. Under pathophysiological conditions, TDP-43 is cleared from the nucleus and accumulates in the cytoplasm [6, 68, 114]. The translocation into the cytoplasm has been considered to be a consequence of impaired nuclear transport and solubility of TDP-43 [159]. This is consistent with findings that seeded aggregation of TDP-43 in cytoplasm may induce the mislocalization of TDP-43 by impairing nuclear transport [17], and that proteins playing a role in nucleocytoplasmic transport are present in pathological TDP-43 aggregates [37].
The neurotoxicity of pathological TDP-43 protein has not been fully clarified. Although the redistribution of TDP-43 from the nucleus to the cytoplasm is generally considered to cause loss of TDP-43 nuclear function [32], some studies have found that nuclear depletion is not necessary for neuronal toxicity induced by ALS-associated mutant TDP-43 [10, 13], and that cytoplasmic mutant TDP-43 has an important role in neurodegeneration [17]. More recent animal model studies support the hypothesis that TDP-43 progression is induced by a gain of toxic function in the cytoplasm, in addition to the loss of nuclear function, and that both are involved in the progression of TDP-43 proteinopathy [50, 51, 57, 138]. Understanding in detail how TDP-43 leads to neurodegeneration will help direct future therapy.
TDP aggregates and their molecular alterations
Biochemical, immunochemical and protein-chemical analyses of materials isolated from the brains of patients with TDP-43 proteinopathies have uncovered the characteristic features of pathological TDP-43. Immunoelectron-microscopic studies demonstrated that the protein accumulates as abnormal fibrils or filaments with cellular organelles, irregularly shaped structures and lipofuscins. The fibrils appear to be amyloid-like, even though they are intracellular proteins, whereas the classical histopathological “amyloid” exists as extracellular proteinaceous deposits exhibiting beta sheet structure [6, 68, 116]. More extensive studies demonstrated that the protein is poorly sarkosyl-soluble, hyperphosphorylated, ubiquitinated, and abnormally cleaved into small fragments (C-terminal fragments) [6, 68, 86, 116]. These data indicate that functional consequences might ensue from the distinctive differences between normal and abnormal TDP-43 (Fig. 2a). It has been suggested that aggregation-prone N-terminal fragments produced by proteolytic cleavage may cause TDP-43 pathologies, but TDP-43 cleavage peptides generated by enzymes such as caspase or calpain have not been detected by nano-flow liquid chromatography–ion trap mass spectrometry [86]. Furthermore, aggregation of full-length TDP-43 precedes generation of TDP-43 C-terminal fragments in the seeded cellular model of TDP-43, suggesting that production of C-terminal or N-terminal fragments is not essential for the formation of intracellular TDP-43 aggregates [86].

Postulated molecular mechanisms of accumulation and propagation of pTDP-43 in human brain. a Normal physiological status and pathophysiological status of TDP-43 in human brain. As shown on the left, TDP-43 primarily resides in the nucleus and is trafficked between the nucleus and cytoplasm in the cells under normal physiological conditions. TDP-43 has roles in regulating gene expression at the transcriptional level, regulating gene splicing, and stabilizing mRNA. Under pathophysiological conditions in patients with pTDP pathology (right), TDP-43 is cleared from the nucleus and accumulates in the cytoplasm. Pathological TDP-43 is fragmented into C-terminal fragments, which are hyperphosphorylated, ubiquitinated and less soluble, and might represent immature products available for aggregation. P phosphorylated, TDP-43 TAR DNA-binding protein 43, U ubiquitinated. b TDP-43 proteinopathy can involve several strains of seeds. Each pTDP-43 strain accumulates in different cell types and regions of the central nervous system. The strains move from one region to another, maintaining their original form. All of these phenomena may contribute to selective local vulnerability. The expression levels of isoforms of the cognate proteins may differ among cells and compartments, thereby promoting or restricting the spread of the seeds and defining the strain of seed that is propagated
As in other neurodegenerative diseases, hyperphosphorylation seems to occur in the aggregated TDP-43 proteins. Many phosphorylation sites, including serine, threonine, or tyrosine residues, have been reported within TDP-43 in cellular experiments, and these residues may have some functional role in TDP-43. However, intensive immunochemical studies using more than 20 phosphorylation-site-specific antibodies for the human TDP-43 sequence revealed that several of the antibodies (pS379, pS403/404, pS409, pS410 and pS409/410) stained the pathological TDP-43 inclusions. These antibodies specifically recognize hyperphosphorylated full-length and cleaved forms of abnormal TDP-43, and thus are frequently used for immunostaining and immunoblotting studies [62, 68]. The phosphorylation and fragmentation were recapitulated in a cellular model of seeded aggregation of TDP-43 [117] (Fig. 2a). In addition, recombinant full-length human TDP-43 forms structurally stable, spherical oligomers that are neurotoxic in vitro and in vivo [50]. In fact, cellular aggregate formation or accumulation of TDP-43 C-terminal fragments is not primarily responsible for the development of the observed disease phenotype in mutant or wild-type TDP-43 mice [43]. These findings also suggest that full-length TDP-43, not the cleavage fragments, participates at an early stage in TDP-43 pathology and also fragmentation might occur after the accumulation of TDP-43 [86].
Notably, patients with PGRN gene (GRN) mutations have high frequency of neuronal intranuclear inclusions (NIIs) containing TDP-43. The formation mechanism of NIIs is still unknown, but one possibility is that the production of NIIs might be associated with the deficiency of PGRN, which is involved in the regulation of lysosomal function since this would lead to the increased lysosomal gene expression and protein levels [144, 145] (Fig. 2a).
Physiological microenvironments associated with neuronal toxicity
Previous research on physiological microenvironments associated with neurodegeneration in TDP-43 proteinopathy has focused on stress granules (SGs). These are membrane-less discrete cytoplasmic structures, containing mRNA and associated proteins that form in cells as a protective response to stress [30]. SGs have long been thought that they contribute to some degenerative diseases, including ALS, FTD, myopathy, and possibly AD and other tauopathies [12, 29, 150]. Many prior studies suggest that TDP-43 and other diseases associated with RNA-binding proteins colocalize with SGs, and SGs might regulate the translational stress response [151]. However, it is still unclear whether TDP pathology arises from SG pathology since colocalization of TDP-43 and SG proteins is hardly observed in human brains [160]. Recent papers demonstrate that cytoplasmic inclusions containing aggregated pTDP-43 can develop through three pathways: direct aggregation or phase-separated intermediates involving ejection from SGs or seeding with exogenous fibrils [58, 105]. These studies suggest that pathological TDP-43 granules are distinct from SGs, although in some cases SG appears to serve as an intermediate. They also indicate that most of the nuclear pore proteins do not colocalize with TDP-43 aggregates, implying that the aggregation occurs through an independent process [58, 105].
TDP-43 expression in non-neuronal cells
In addition to neurons, TDP-43 is abundantly expressed in glia, as well as many other cell types. Oligodendrocytes with accumulations of pTDP-43 frequently appear in FTLD/ALS brains [5, 26], and TDP-43-positive inclusions in astrocytes have been recognized in Alexander’s disease [152]. Basically, microglia play a role in mediating neurotoxicity and neuroprotection, and maintaining homeostasis, including control of the formation of synapses, refining neuronal circuits, eliminating redundant synapses, and healing responses to injury [139].
Recent studies of mice with TDP-43 pathology induced with TDP-43 from patients with TDP-43 proteinopathy established that white matter oligodendrocytes and astrocytes are capable of developing phosphorylated TDP-43 aggregates [122], and abundant reactive microglia selectively cleared neuronal human TDP-43 [74]. Studies in animal models of other proteinopathies, such as α-synuclein, have also described neurodegeneration with inflammation, and activated microglia [136]. These findings suggest that inclusions or degenerating neurons with aggregates may be cleared by microglial phagocytosis, which may account for the neuroprotective action of microglia [136, 139]. Modulation of microglia-mediated function during the progression of TDP-43 pathology may have potential as a new therapeutic strategy.
Phenotypic diversity in TDP-43 proteinopathy
Clinical phenotypic diversity in TDP-43 proteinopathy
Many neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS), are classified according to their clinical symptoms and neuropathological features. Indeed, major clinical symptoms correspond well to the anatomical brain regions showing neuronal loss with reactive gliosis. These pathological changes are associated with the accumulation of abnormal proteins with conformational changes and modifications, which occur intracellularly in neurons or glial cells, or extracellularly [89].
ALS and FTLD-TDP are sporadic and familial neurodegenerative diseases characterized neuropathologically by cellular aggregates of TDP-43. The differences in TDP-43 pathology are associated with differences in clinical manifestations, and ALS and FTLD are generally diagnosed on the basis of clinical symptoms. However, some patients may show symptoms common to both diseases. For example, clinical overlap between ALS and behavioral variant frontotemporal dementia (bvFTD) has been recognized, with up to 50% of ALS patients showing some executive function deficits [98, 127]. Also, up to 15% of bvFTD patients show motor neuron dysfunction [34, 127].
There are three major clinical phenotypes of frontotemporal lobar degeneration (FTD): bvFTD, semantic variant primary progressive aphasia (svPPA) and nonfluent/agrammatic primary progressive aphasia (naPPA) [36, 44, 48]. Each clinical syndrome among these diseases is associated with topographically distinct cerebral involvement, based on pathological protein deposits. bvFTD is associated with symmetrical (though sometimes right-sided) frontotemporal dysfunction, svPPA with left anterior temporal deficits and naPPA with left frontotemporal dysfunction (Fig. 3).

Specific topographic patterns of atrophy in FTLD-TDP. bvFTD predominantly shows frontal lobe atrophy, svPPA shows left anterior temporal tip atrophy, and naPPA shows left frontotemporal atrophy. Each clinical syndrome is associated with distinct cerebral involvement behavioral variant FTD: bvFTD, semantic variant PPA: svPPA, nonfluent/agrammatic PPA: naPPA
Loss of empathy, apathy, and selfishness are more frequent features of bvFTD, but may be found commonly in all subtypes [18]. These symptoms are associated with atrophy and dysfunction of the right anterior cingulate cortex and superior frontal gyrus, while disinhibition is associated with the right subgenual cingulate cortex and orbitofrontal cortex [121, 163], overeating with an orbitofrontal striatal circuit [161], and executive dysfunction with the dorsolateral and prefrontal cortex [73]. Fluent speech, progressive impairment of single-word comprehension, preserved articulatory abilities, and dysfunction of semantic memory are features of svPPA [63]. Patients with naPPA show apraxia of speech or expressive agrammatism: single-word comprehension and object knowledge are relatively preserved, and behavioral symptoms are less common in comparison with their high frequency in svPPA. Cognitive decline, dementia, and alterations in high-order brain functions are associated with the involvement of the limbic region, including the entorhinal cortex and hippocampus, and neocortical areas. Most svPPA can be classified into TDP proteinopathy, whereas bvFTD and naPPA are correlated with FTLD-tau [137].
TDP-43 pathology in aged brains and other neurodegenerative diseases
In FTLD-TDP and ALS, neuronal loss, reactive gliosis, microglial activation and superficial laminar spongiosis are seen in the neocortex and hippocampus [48]. TDP-43 pathology is present throughout the CNS with relative preservation of the occipital cortex and cerebellum [61]. Subcortical areas, including the nucleus accumbens, basolateral amygdala and caudate/putamen, are also progressively affected [61]. In movement disorders such as ALS, the basal ganglia, thalamus, brainstem nuclei, cerebellar cortex and nuclei, motor cortical areas and lower motor neurons of the spinal cord are involved. These findings suggest that selected neuronal populations may be particularly vulnerable to TDP-43 aggregates. TDP-43 immunoreactive structures include neuronal cytoplasmic inclusions (NCIs), neuronal pre-inclusions, dystrophic neurites (DNs), neuronal intranuclear inclusions (NIIs), and glial cytoplasmic inclusions (GCIs).
The deposited protein is hyperphosphorylated, ubiquitinated and abnormally cleaved to generate C-terminal fragments [6, 68, 116]. The phosphorylation sites of abnormal TDP-43 are located at Ser379, Ser403, Ser404, Ser409 and Ser410. TDP-43 inclusions are thioflavin-S positive [22], suggesting that TDP-43 is accumulated as amyloid-like fibrils.
TDP-43-positive NCIs are frequently found in the frontotemporal neocortex and dentate granule cells of the hippocampus in ALS, FTLD and FTLD–MND [76, 118, 157]. They are also positive for ubiquitin and p62, but negative for tau, α-synuclein, amyloid beta and FUS [6, 36, 44, 116, 118]. Skein-like and spherical or round inclusions are present in motor neurons in ALS. These inclusions are detected in 100% of TDP proteinopathy brains. Diffuse or granular or dash-like cytoplasmic inclusions, which are dispersed throughout the neuronal somatodendritic domain, are found in motor neurons of cranial nerve nuclei or spinal cord of ALS and FTLD-TDP [44, 60]. These inclusions are considered to be in a transitional state and are referred to as “pre-inclusions”, since they are negative for ubiquitin and p62 [5]. As for DNs, two types of DNs, short DNs and elongated DNs, have been recognized [36, 71]. Both types of DNs are typically most numerous in layer II of the frontal and temporal cortices, although the elongated DNs are generally more widely dispersed throughout the entire cortex in comparison with the short DNs [71]. NIIs show a lentiform or “cat’s eye” appearance and are present in small neurons in multiple neuroanatomical sites. They appear frequently in familial cases [60]. GCIs with coiled body-like or globular morphologies are frequently found in the cerebral white matter and the spinal cord in ALS and FTLD–MND [27, 71, 115]. GCIs are positive for TDP-43 and p62, but are mostly negative for ubiquitin [9, 60]. Most GCIs are considered to be of oligodendrocytic origin on the basis of double immuno-labeling for pTDP-43 and a complement protein, C4d [7] (Fig. 4).

Pathological features of TDP-43-positive structures. Macroscopic photograph of an FTLD-TDP patient’s brain. The left hemisphere shows severe atrophy in the frontal cortex and mild atrophy in the temporal tip (a). TDP-43-positive structures detected in patients with TDP proteinopathy. The dentate gyrus of the hippocampus shows neuronal cytoplasmic inclusions (NCI) with no nuclear staining (b). Rounded NCI in the entorhinal cortex (c), skein-like inclusions in the lower motor neurons (d), and inclusions with granular appearance in the amygdala (e) can be seen. There are dystrophic neurites (DNs) in the temporal cortex (f). Neuronal intranuclear inclusions with a “cat’s eye” shape are present in the entorhinal cortex (g) and glial cytoplasmic inclusions are seen in temporal white matter (h). b–h Stained with phosphorylated TDP-43-specific antibody (pS409/410). Scale bars: a 1.5 cm; b, d, f 20 μm; c, e, g 10 μm
TDP-43 pathology is typically classified into four subtypes (types A–D) in FTLD-TDP, based on the predominant type of TDP-43-positive structures and their distribution [101]. Type A is characterized by numerous short DNs and crescentic or oval NCIs; type B shows NCIs throughout all cortical layers with few DNs; type C presents a predominance of elongated DNs in upper cortical layers with few NCIs; and type D is characterized by numerous short DNs and frequent lentiform NIIs (Figs. 2b, 5a). Recently, a new type, type E, was identified, in which granulofilamentous neuronal inclusions, abundant grains, and oligodendroglial inclusions were prominent [95].

Pathological and biochemical classification of TDP proteinopathy. a FTLD-TDP subtypes A to C. Type A is characterized by numerous neuronal cytoplasmic inclusions (NCIs) and short dystrophic neurites (DNs). This type frequently has neuronal intranuclear inclusions (NII) in the affected regions; type B shows numerous NCIs; type C has long and tortuous DNs. b Immunoblots of insoluble TDP-43 from patients with TDP proteinopathy, probed using anti-phosphorylated TDP-43 antibody (pS409/410). The band patterns of TDP-43 C-terminal fragments from the three TDP-43 types are different (shown schematically on the left). Type B has three major bands at 23, 24 and 26 kDa and two minor bands at 18 and 19 kDa (lane 2). Type C has two major bands at 23 and 24 kDa and two minor bands at 18 and 19 kDa (lane 3). Type C has an intense band at 23 kDa, whereas type B has a strong 24 kDa band. The band pattern of type A seems intermediate between types B and C (lane 1). Molecular weight markers of migrated protein standards are shown in kDa. Scale bar: 20 μm
TDP pathology should be evaluated from multiple points of view. Neuropathological comorbidity has recently attracted attention. TDP-43 pathology frequently appears in other neurodegenerative diseases, including AD, argyrophilic grain disease (AGD), dementia with Lewy bodies (DLB), chronic traumatic encephalopathy (CTE), hippocampal sclerosis (HS), and Guam ALS [8, 61, 67, 90, 109, 113, 162]. Concomitant TDP-43 pathology has a significant impact on the clinical features of AD [82, 83]. TDP-43 pathology in AD has been classified into six stages, supported by correlations with neuroimaging and clinical features [82, 83]. It begins with the amygdala (stage I), then spreads into the subiculum and entorhinal cortex (stage II), and involves the dentate gyrus and occipitotemporal cortex (stage III). Next, the insular, ventral striatum, basal forebrain and inferior temporal cortex are affected (stage IV). Then it appears in the substantia nigra, inferior olive nucleus, and midbrain tectum (stage V), and finally in the basal ganglia and middle frontal cortex (stage VI) [82, 83]. Progression is associated with impaired cognition, memory loss, and medial temporal atrophy in AD [83]. Concomitant TDP-43 pathology was also found in 53–60% of DLB cases, 60% of AGD cases (Fig. 3e) and 100% of Huntington’s disease cases [8, 57, 130]. TDP pathology was seen in 89.9% of patients with HS compared with 9.7% of patients without HS [113]. TDP-positive inclusions and neurites were also found in 36–85% of CTE cases, ranging from early to late stage [109].
TDP-43 pathology is present in a high proportion of cognitively normal elderly persons [11]. In cases of primary age-related tauopathy (PART), which is characterized by the presence of neurofibrillary tangles (NFTs) in the limbic and brain stem region and absent-minimal β-amyloid deposition [41], 26.7% have TDP-43 pathology and the presence of TDP-43 was associated with significantly greater amygdala, hippocampal, and anterior temporal atrophy [80].
As we described above, TDP-43 pathology frequently occurs in AD and DLB cases. In 20–30% of these cases, the morphological pattern of cortical TDP-43 immunoreactivity and the band pattern of C-terminal fragments of insoluble TDP-43 on immunoblots are similar to those of type A FTLD-TDP, which is seen in cases with GRN mutation [8].
Another study classified TDP-43 pathology in non-FTLD brains into two subtypes, TDP type-α and TDP type-β. The former shows typical TDP-43 immunoreactive inclusions, while the latter is characterized by the presence of TDP-43 immunoreactivity adjacent to/associated with NFTs in the same neuron [81]. The age at death of type-α was older than that of type-β (median 89 years vs. 87 years). Hippocampal sclerosis was present in 60% of type-α patients and 15% of type-β patients. A pattern of widespread TDP-43 deposition commonly extended into the temporal, frontal and brainstem regions in type-α, whereas TDP-43 deposition was predominantly located in limbic regions, including amygdala, entorhinal cortex and subiculum of the hippocampus in type-β [81]. The frequency of TMEM106B protective (GG) and risk (CC) haplotypes (SNP rs3173615 encoding p.T185S) is high in type-α compared to type-β in non-FTLD-TDP cases [81] (Fig. 6).

Clinical, pathological, and genetic spectra of TDP proteinopathy. ALS amyotrophic lateral sclerosis, bvFTD behavioral variant frontotemporal dementia, MND motor neuron disease, naPPA nonfluent/agrammatic PPA, svPPA semantic variant PPA, GRN granulin, VCP valosin-containing protein
Recently, the new term limbic-predominant age-related TDP-43 encephalopathy (LATE) was proposed to describe the presence of TDP-positive lesions in AD, as well as in older adults [112]. LATE neuropathological change (LATE-NC) is defined by a stereotypical TDP-43 proteinopathy in older adults, with or without coexisting hippocampal sclerosis pathology, and is distinguished from FTLD-TDP. LATE-NC is also a common TDP-43 proteinopathy associated with an amnestic dementia syndrome similar to AD. LATE includes already characterized phenotypes, but involves no new TDP-43 pathologic subtype, no link between TDP pathology with new cognitive symptoms, and no characteristic biochemistry [79]. Although the term aims to encourage future research, as well as development of neuroimaging and molecular biomarkers, the concept may be confusing in relation to AD and should be considered further.
Genetic predisposition leading to differential vulnerability
The pathological classification of TDP-43 proteinopathy is supported by clinical and genetic correlations. svPPA is associated with type C, and FTLD-MND or clinical signs of MND with type B, and naPPA with type A, although bvFTD is not as strongly correlated with any special pathological type [21, 78, 85, 100]. Most cases of ALS/FTLD are sporadic but ~ 5% is familial. Regarding the genetics, clinical clusters of FTD and ALS have been reported to be associated with several genes, including granulin precursor (GRN) [15, 42], valosin-containing protein (VCP) [153], TAR DNA-binding protein (TARDBP) [85, 91], chromosome 9 open reading frame 72 gene (C9orf72) [45, 100, 124], sequestosome 1 (SQSTM1) [148], ubiquilin 2 (UBQLN2) [46], TANK-binding kinase 1(TBK1) [56], T cell-restricted intracellular antigen-1 gene (TIA1) [103], cyclin F (CCNF) [158], coiled-coil-helix-coiled-coil-helix domain-containing protein 10 (CHCHD10) [16] and optineurin (OPTN) [35]. Superoxide dismutase 1(SOD1) has also been linked with ALS [128]. C9orf72 repeat expansion is the most frequent genetic factor in sporadic and familial cases [2, 33, 66], but 40% of familial cases have no established genetic cause, and unknown genes may be involved [2, 66]. Cases with a C9orf72 expansion repeat mutation have additional p62-positive and TDP-43-negative NCIs, which are composed of dipeptide repeat proteins (translated from the C9orf72 expansion repeats) [100]. Psychotic symptoms, such as delusions and hallucinations, are likely to present in C9orf72 repeat expansion carriers [47, 138], although they are generally rare in FTD [97]. Also, family members of patients carrying C9orf72 repeat expansion might have high incidences of psychiatric illness [47]. GRN is mostly associated with type A, and C9orf72 with type B, or less commonly with type A [100, 101]. Associated genes are still unknown in types C and E [95]. Type D has been reported to be highly associated with inclusion body myopathy with early-onset Paget disease of bone and frontotemporal dementia (IBMPFD) caused by VCP mutations. Missense mutations in VCP affect various cellular processes, including ubiquitin-dependent protein quality control, cell division, and nuclear envelope formation [110]. As a result, the consolidation of aggregate-prone proteins into inclusion bodies via multivesicular body formation is induced and causes the pathogenic features [149].
Various related genes, apolipoprotein E (APOE), granulin (GRN), transmembrane protein 106B (TMEM106B), ATP-binding cassette sub-family member 9 (ABCC9) and potassium channel subfamily M regulatory beta subunit 2 (KCNMB2), have been reported to be risk alleles associated with pathological manifestations in FTLD-TDP and LATE [112].
Progression pattern of TDP-43 pathology in human brains
Clinical phenotypes are associated with characteristic distribution patterns of the pathology in the course of disease progression. The progression pattern of TDP-43 pathology has been identified [26, 27]. In bvFTD, pTDP-43 pathology progresses in four distinct topographic stages [26]. Stage I involves the orbital gyri, gyrus rectus, and amygdala, and stage II progresses to the middle frontal and anterior cingulate gyrus as well as anteromedial temporal lobe areas, the superior and medial temporal gyri, striatum, red nucleus, thalamus, and precerebellar nuclei. Stage III shows involvement of the motor cortex, bulbar somatomotor neurons, and the spinal cord anterior horn, whereas finally, stage IV shows the highest burden of pathology in the visual cortex [26]. bvFTD is associated with all subtypes of TDP pathology. In the case of svPPA with type C, the progression pattern of TDP-43 pathology is not yet established.
The progression pattern of pTDP-43 in ALS patients has also been characterized [24, 27]. ALS with TDP deposition begins in the upper motor neurons in the cortex, lower motor neurons in the spinal cord and lower brainstem (stage I). Stage II progresses gradually to posterior frontal and anterior parietal regions, brainstem reticular formation, and red nucleus. Stage III involved the anterior frontal and basal forebrain, striatum, thalamus (mediodorsal/lateral thalamus), and substantia nigra. Finally, patients show involvement of the anterior temporal lobe including hippocampus (stage IV) [24, 27] (Fig. 7).

Progression of TDP pathology in bvFTD and ALS The upper figure shows TDP-43 pathology progression in behavioral variant-frontotemporal dementia (bvFTD) of Types A and B, possibly propagating along axonal pathways. Stage 1: pTDP-43 pathology begins with neurons and oligodendrocytes of the basal and anterior portions of the prefrontal neocortex and amygdala. Stage 2: the pathology appears caudal to the frontal lobe, including the middle frontal gyrus, insular cortex, and anterior cingulate gyrus, and also in anteromedial areas, including the hippocampus (H), the caudate nucleus and putamen (C/P), and the mediodorsal nucleus (MD) of the thalamus (T), red nucleus (R), precerebellar nuclei (PC), and dorsomedial medullary regions. Stage 3: TDP pathology is increased compared to the first two stages and has spread further into the cortical, brainstem, and spinal motor regions. Stage 4 cases with the highest burden of pathology, showing pTDP-43 deposits in the occipital neocortex (visual cortex, Brodmann 17, 18) Amyotrophic lateral sclerosis (ALS) with TDP-43 pathology is shown in the lower figures. Stage 1: pTDP-43 deposition begins (stage 1) in the agranular motor cortex, or lower motor neurons in the spinal cord and lower brainstem. Stage 2: increasing burden of TDP-43 pathology, with progression to the posterior frontal and anterior parietal regions, brainstem reticular formation (RF), PC and R. Stage 3: TDP pathology involves the anterior frontal and basal forebrain, the C/P, MD and lateral thalamus (LT), and substantia nigra (SN), Stage 4: TDP pathology has spread into the anterior temporal lobe, including the H. cornu ammonis subregions 1 and 2 of the hippocampus (CA1–2), hypoglossal nucleus (XII), and inferior olivary complex (IOC)
Methods for assessing clinical phenotypic diversity
Several methods have been developed with the aim of identifying the differences between TDP-43 and other proteinopathies. Many researchers have investigated TDP-43 in cerebrospinal fluid (CSF) since it is considered to reflect the TDP pathology in human brain. However, the results are conflicting. One report found that ALS and FTLD patients had higher TDP-43 levels in CSF, compared to control patients [141], but another report indicated that TDP-43 CSF levels were higher in ALS, in comparison with FTLD [84]. In the latter paper, there was a difference in C9orf72 mutation between ALS and FTLD patients, but no difference was detected between carriers and non-carriers. Furthermore, a non-significant trend of lower pTDP-43 levels in ventricular CSF was reported in FTLD-TDP compared to controls [92]. These results suggest that TDP-43 CSF level does not adequately reflect TDP-43 pathology, and may not be a useful biomarker [140]. Further, TDP-43 analysis in blood is not yet available for the diagnosis of TDP-43 pathology, due to the lack of sufficiently sensitive quantification methods [53, 54]. However, more advanced methods should be developed in the near future.
Several neuroimaging studies of FTLD-TDP and ALS have been reported. Volumetric structural MRI scans, cortical thickness analyses, diffusor tensor imaging, and to a lesser extent functional techniques, including functional MRI and FDG-PET have been utilized to aid diagnosis in clinical practice [20, 33]. Generally, patients with FTLD classically show frontal and temporal atrophy, involving the orbitofrontal/dorsolateral prefrontal cortices and the temporal poles, and hypometabolism in these regions is often asymmetrical. As mentioned above, there are characteristic patterns of atrophy in the three clinical phenotypes of FTLD. MRI or positron emission tomography (PET) findings have revealed frontal and/or anterior temporal atrophy or hypometabolism in bvFTD [123], left posterior frontoinsular atrophy, hypoperfusion or glucose hypometabolism in naPPA, and predominant anterior temporal lobe atrophy or hypoperfusion or hypometabolism in svPPA [64, 129]. In the diagnosis of ALS, imaging is used only for exclusion of other diseases [28] (Fig. 8).

Imaging of an FTLD-TDP patient showing the clinical presentation of naPPA. Left photo shows T1-weighted MRI at onset (a) and 2 years after onset (b). Frontal and temporal atrophy on axial slices (two photos on the left) is prominent. Coronal slices (two photos on the right) indicate atrophy in the dorsolateral prefrontal cortex and medial and inferior temporal cortex with enlargement of the temporal horn. Single-photon-emission computed tomography using technetium-99 m ethyl cysteinate dimer (ECD-SPECT) revealed a reduction in the bifrontal regions (Right photo)
Imaging studies indicate that neurodegenerative diseases are caused by degeneration within specific intrinsic functional connectivity networks vulnerable to their pathologies [132, 165]. fMRI studies in bvFTD show attenuated connectivity within an anterior network of dorsal anterior cingulate and frontoinsular cortices connecting to subcortical and limbic structures [131, 166] and network disruption, with reduced mean network extent and increased path length, compared with healthy controls [1]. These results enable us to recognize sites of early disruption as vulnerable regions, and also throw light on the connectivity of the areas involved in the diseases. Further molecular pathological approaches will help to establish which aberrant proteins progress along networked brain structures, and whether specific pathological subtypes can be linked to specific forms of neural network degeneration [52].
Since the discovery of C9orf72 in 2011, the role of the repeat expansion in TDP-43 proteinopathy has been investigated by means of imaging techniques. Recent neuroimaging analyses demonstrate that patients with C9orf72-positive bvFTD show symmetric atrophy most prominently in the anterior insula, anterior cingulate, and frontotemporal cortex, in keeping with the sporadic bvFTD-associated pattern [23, 75, 104, 133, 155]. Some studies indicate that C9orf72 carriers exhibit atrophy of the parietal lobe [75, 155] and occipital lobe [133, 155], which are not typically involved in bvFTD. A resting-state functional MRI study found that patients with bvFTD with or without C9orf72 expansion show convergent large-scale network breakdowns despite having distinctive atrophy patterns [96]. In ALS, extensive cortical and subcortical frontotemporal involvement has been reported in association with the C9orf72 genotype, compared to the relatively limited extramotor pathology in patients with C9orf72-negative ALS [19]. The changes in the orbitofrontal, fusiform, thalamic, superior temporal regions, and Broca area, and posterior cingulate in ALS are distinct from those seen in the C9orf72 genotype [19]. As for PET, C9orf72-positive ALS had a more widespread central nervous system involvement than C9orf72-negative ALS, with or without FTD comorbidity [38].
Strains and phenotypic diversity
Selective neuronal and regional susceptibility
Understanding the underlying causes is crucial for building effective treatment strategies. As described, neurodegeneration in ALS selectively involves the upper and lower motor neurons within the pyramidal motor system [134]. Motor neurons are different from many other neuronal cells in both size and shape. In addition, characteristic anatomical features include a large cell body with a high level of mitochondrial activity and a long axonal process with a high content of neurofilament proteins [134]. Those anatomical features are associated with mRNA splicing, oxidative stress, proteosomal and mitochondrial dysfunction, and glutamatergic toxicity with damage to critical target proteins in the pathogenesis of ALS [65, 134]. Also, axonal cytoskeletal disorganization has been considered as a conspicuous feature of ALS. ALS linked with mutations in TDP-43 shows impairment of the axonal transport of RNA granules in animal models of TDP-43 proteinopathy [3].
TDP-43 accumulations are detected in unaffected cells in ALS, though some Betz cells, which mainly degenerate in ALS, are known to lack TDP-43 accumulation [25, 59]. This may imply that there is no direct correlation between the presence of the accumulations and cell death. The Betz cells in upper motor neurons of patients display a distinct set of intracellular defects, especially at the nuclear membrane, mitochondria and endoplasmic reticulum [59]. Another argument against the hypothesis is that only cells with TDP-43 accumulation cause degeneration. Previous data suggest that MND with ALS-linked mutants producing loss and gain of splicing function of selected RNA targets at an early disease stage does not require TDP-43 accumulation and loss of TDP-43 from the nuclei [10].
Many physiological and pathological studies indicate system degeneration in ALS and FTLD. ALS patients present focal clinical symptoms showing upper or lower motor neuron predominance and clear alterations in motor function in one body region at a specific time, typically within a timescale of weeks to months [142]. The neuroanatomical distribution of pTDP-43 pathology allowed classification of FTLD cases into several clinical subtypes, as described above. Thus, the disease does not progress randomly across contiguous anatomical pathways [142]. It is important to consider the pattern of progression of neurodegeneration at the macroscopic level, in addition to the selective neuronal vulnerability at the microscopic level.
The vulnerable circuits and cell types in FTD have recently been investigated. Especially in bvFTD, von Economo neurons and fork cells within the anterior cingulate and ventral anterior insular cortices are affected in early degeneration [88, 111]. The large layer 5 projection neurons in the regions undergo early dropout in bvFTD [88, 111]. In addition, the presence of TDP-43 inclusions was associated with striking nuclear and somatodendritic atrophy and the inclusion fraction in those cells correlated with symptom severity, such as loss of emotional empathy, in bFTD [111]. These insights regarding selective vulnerability and the mode of their progressive degeneration might lead to the identification of new targets for therapy and effective treatments for numerous neurodegenerative diseases.
Current evidence regarding strains and progression
The different banding patterns of abnormal TDP-43 fragments in ALS and FTLD may represent different TDP-43 strains with different conformations. Biochemical analysis of different brain regions and spinal cord in individual patients with TDP-43 proteinopathy revealed three distinct C-terminal banding patterns [40, 68, 147]. This strongly suggests that the same form of abnormal TDP-43 molecule is consistently deposited in different brain regions in each patient. It is unlikely that the same conformational change would occur synchronously in different brain regions, and it seems more likely that abnormal protein produced in cells is transferred to different regions, transmitted from cell to cell and propagated in vivo [40, 68, 69, 72, 147]. In addition, novel cell-based techniques indicate that there are distinct forms of pathological TDP-43 with different biochemical features, which correlate with the FTLD subtypes; they manifest distinct neurotoxicities and show different seeding activities in culture [40, 68, 72].
In prion diseases such as CJD and bovine spongiform encephalopathy, the different patterns of protease-resistant bands can be used to identify the etiology of the diseases [40, 120]. For example, protease-resistant prion from new variant CJD exhibits a different banding pattern from that in sporadic CJD cases, and the band pattern was indistinguishable from that of mice infected with bovine spongiform encephalopathy prion. These observations are consistent with the idea that the different banding patterns of abnormal TDP-43 fragments in ALS and FTLD might represent different TDP-43 strains with different conformations.
A hypothesis that has received much attention recently is that neurodegeneration might spread from cell to cell by a mechanism that is seen in prion diseases [117]. According to this hypothesis, abnormal insoluble TDP-43 in brains of patients exhibits prion-like properties. Recently, pathological TDP-43 derived from FTLD-TDP brains was shown to induce formation of de novo TDP-43 pathology with subsequent spreading throughout the central nervous system in a region- and time-dependent manner in experimental animal models [122]. It was also reported that TDP-43 could be released from cells via secreted vesicles called exosomes. This could facilitate the propagation of prion-like TDP-43 aggregates from one cell to others. The types of TDP-43 aggregates (strains) initially generated in neuronal/glial cells spread to the neighboring cells and determine the TDP-43 pathologies. One study found that inhibition of exosome secretion exacerbated the disease phenotypes of transgenic mice expressing human TDP-43 mutant [74]. Morphological analyses of mouse primary cortical neurons, patients’ fibroblasts, and stem cell-derived neurons showed that aggregation of TDP-43 triggered the sequestration and mislocalization of nucleoporins and transport factors, and interfered with nuclear protein import and RNA export [37].
Exogenous and environmental factors modulating selective regional vulnerability
Clinical studies suggest that traumatic brain injury (TBI) might be associated with an elevated risk of ALS, as well as other devastating neurodegenerative diseases [125, 126]. Chronic traumatic encephalopathy (CTE) is a neurodegenerative disease caused by repeated TBI associated with contact sports, and diseased brains contain abnormal accumulations of hyperphosphorylated tau [106]. Head trauma induces focal axonal injury, microhemorrhage and gliosis in close proximity to the affected area, then p-tau accumulates in neurons in the perivascular regions due to axonal injury, breach of the blood–brain barrier, and neuroinflammation, and finally p-tau spreads throughout the brain [108]. A previous paper describes patients with repetitive head trauma who showed widespread TDP-43 inclusions in the CNS, including motor neurons [107]. A study in Drosophila models of ALS indicated that repetitive trauma promotes the accumulation of TDP-43, with SGs in the brain, and also exacerbates neurodegenerative phenotypes as indicated by mortality and locomotor dysfunction [4]. Reversible induction of TDP-43 in cortical neurons after traumatic injury was also observed in a mouse stab wound model of TBI [156]. A single instance of TBI might not necessarily cause TDP progression, but repetitive head trauma may initiate or modify the onset or progression of ALS [146]. The precise etiology remains unclear and further research is needed to elucidate the impact of trauma on TDP-43 propagation.
In terms of selective vulnerability, exposures outside the CNS may influence motor neurons themselves. Especially in the spinal cord, motor neurons lack the protection of the blood–brain barrier at the axon termini. The neuromuscular junction seems vulnerable and the retrograde transport of aberrant factors may be able to occur at this site in patients [142].
Experimental modeling of phenotypic diversity
The development of multiple experimental models of TDP-43 proteinopathy is essential to clarify the pathogenic mechanism underlying TDP-43 propagation. Protease-resistant banding patterns of pathological insoluble TDP-43 are useful for biochemical classification of the subtypes [147]. Cell-to-cell transmission of TDP-43 aggregates also occurs in cell culture, and exosomes are likely to be involved in pathological TDP-43 propagation [51, 117, 135]. SH-SY5Y cells treated with insoluble TDP-43 extracted from ALS or FTLD-TDP brains expressed aggregates of phosphorylated and ubiquitinated TDP-43 in a self-templating manner [117]. Immunohistochemical analyses established that the C-terminal fragments of each disease type acted as seeds in these cells, inducing seed-dependent aggregation of TDP-43 [117]. The seeding ability of insoluble TDP-43 was hardly affected by protease treatment, but was abrogated by formic acid. Another study indicated that anterograde and retrograde transport of TDP-43 oligomers occurs in cultured cells, providing support for the idea of cell-to-cell transmission of pathological TDP-43 [51]. Furthermore, residues 274–353 are essential for the conversion of TDP-43 to amyloid-like fibrils, and aggregation of TDP-43 by seeding with different peptides induces various types of TDP-43 pathologies [135]. These findings indicate that insoluble TDP-43 has prion-like properties that likely play a role in disease progression of patients with TDP-43 proteinopathy.
As in the cases of tau and α-synuclein, the prion-like propagation of TDP-43 protein in vivo has been investigated by injecting brain extracts from aged transgenic mice showing pathology, or synthetic protein fibrils, or extracts from brains of patients into mouse brains. Animal models of TDP-43 propagation are not easy to prepare, compared to models of tau and α-synuclein propagation. It is very difficult to prepare substantial amounts of natively folded recombinant proteins or fragments in E. coli, and so it is difficult to form synthetic amyloid-like TDP-43 fibrils [70]. In addition, unlike α-synuclein or tau, TDP-43 is localized in the nucleus and its expression is tightly self-regulated [14], which may stabilize the conformation of TDP-43 in vivo and prevent the formation of abnormal TDP-43 pathology in non-Tg mice. However, more recent studies succeeded in the formation of TDP-43 pathology with subsequent spreading throughout the central nervous system in a region- and time-dependent manner by injecting pathological TDP-43 of FTLD-TDP into transgenic CamKIIa-hTDP-43NLSm mice, which overexpress mutant TDP-43 lacking a nuclear localization signal [122]. The propagation of TDP-43 pathology in the injected mice resulted in widespread cortical, hippocampal, and subcortical pTDP-43 accumulation in both ipsilateral and contralateral hemispheres, consistent with cell-to-cell transmission of TDP-43 pathology following the neuroanatomical connectome from the injection site. Another report clarified phenotypic heterogeneity among mutant mice and identified distinct transcriptomic profiles corresponding to differing phenotypes. The authors observed changes linked with improved behavior in TDP-43 knock-in mouse with a human TARDBP gene, associated with downregulation of two known modifiers of neurodegeneration, Atxn2 and Arid4a, and upregulation of myelination and translation genes. Identifying the environmental factors that influence the delicate balance in the transcriptome of the brain might be a promising topic for future studies [154]. Other physiological models indicate that aggregation and nuclear depletion are dispensable for neurodegeneration [55, 154]. Further work is necessary to establish the relationship between the seeding activity or strain-like properties of pathological TDP-43 in patients and the neuropathological phenotypes of the mice.
Concluding remarks
Increasing evidence indicates that ALS/FTLD-TDP disease progression is induced by cell-to-cell propagation of intracellular aggregated proteins. Extensive biochemical and biophysical studies needed to clarify the pathomechanisms leading to the presence of distinct pathogenic TDP-43 strains in patients’ brains. Using genome editing technology, it should be possible to generate large knock-in animal models or human iPSCs to overcome overexpression, off-target, and mosaic effects, which could offer promising approaches for therapy [39]. In addition, cryo-electron microscopy (cryo-EM) will be a promising method to clarify the strains of pathological TDP-43. As for tau, a recent report has revealed the structures of the disease-specific folds in the ordered cores of tau filaments, establishing the existence of molecular conformers [49].
Genome-wide association studies have uncovered over 100 loci that are linked with ALS/FTLD [77]. Some ALS/FTLD-TDP patients have a familial history of neurological and/or psychiatric diseases that might possibly be associated with unknown genetic mutations. [14] Identifying those mutations would be helpful to identify the factors modulating disease progression and to provide targets for development of therapeutic interventions. Gene therapy or antisense oligonucleotide (ASO) therapy to knock out a relevant gene or to reduce expression of the protein might also be effective to prevent protein aggregation and prion-like propagation.
Recent years have seen a rapid development of fluid biomarkers for FTLD. In C9orf72 mutation carriers, the concentration of poly GP, which is produced as an abnormal dipeptide repeat protein, could be a target biomarker, in addition to TDP-43 [164]. The development of an immunoassay method for its detection in CSF will be needed.
Identification of imaging biomarkers, such as amyloid-imaging probes, that predict future clinical syndrome will be critical for the identification of suitable candidates for clinical trials. In addition, clinicopathological studies of TDP-43 are expected to provide deeper insights into disease mechanisms, although we should pay attention to the limitations involved in investigating patients with end-stage disease. Such studies should provide further support for the idea that clinical heterogeneity in patients is associated with the heterogeneity of TDP-43 neuropathology resulting from different TDP-43 strains.
Small molecular compounds that can pass through the blood–brain barrier, enter the cells, and bind to intracellular aggregates of abnormal proteins would be promising candidates for pharmacotherapy of neurodegenerative diseases by promoting clearance of pathological TDP-43 proteins, e.g., by activating degradation or protein quality-control systems. Antibody therapy may also be feasible.
In conclusion, combinations of behavioral, functional, and physiological approaches to study the changes in the neuroanatomical system will be crucial for the development of future therapy. In addition, the clinically stereotyped nature of the disease progression, and neuronal brain imaging studies, suggests that a system level vulnerability is a significant aspect of the biological underpinning of TDP-43 proteinopathy.
References
Agosta F, Canu E, Valsasina P, Riva N, Prelle A, Comi G et al (2013) Divergent brain network connectivity in amyotrophic lateral sclerosis. Neurobiol Aging 34:419–427. https://doi.org/10.1016/j.neurobiolaging.2012.04.015
Al-Chalabi A, Hardiman O (2013) The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol 9:617–628. https://doi.org/10.1038/nrneurol.2013.203
Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW et al (2014) Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81:536–543. https://doi.org/10.1016/j.neuron.2013.12.018
Anderson EN, Gochenaur L, Singh A, Grant R, Patel K, Watkins S et al (2018) Traumatic injury induces stress granule formation and enhances motor dysfunctions in ALS/FTD models. Hum Mol Genet 27:1366–1381. https://doi.org/10.1093/hmg/ddy047
Arai T (2014) Significance and limitation of the pathological classification of TDP-43 proteinopathy. Neuropathology 34:578–588. https://doi.org/10.1111/neup.12138
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H et al (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. https://doi.org/10.1016/j.bbrc.2006.10.093
Arai T, Hasegawa M, Nonoka T, Kametani F, Yamashita M, Hosokawa M et al (2010) Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology 30:170–181. https://doi.org/10.1111/j.1440-1789.2009.01089.x
Arai T, Mackenzie IR, Hasegawa M, Nonoka T, Niizato K, Tsuchiya K et al (2009) Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol 117:125–136. https://doi.org/10.1007/s00401-008-0480-1
Arai T, Nonaka T, Hasegawa M, Akiyama H, Yoshida M, Hashizume Y et al (2003) Neuronal and glial inclusions in frontotemporal dementia with or without motor neuron disease are immunopositive for p62. Neurosci Lett 342:41–44
Arnold ES, Ling SC, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D (2013) ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci USA 110:E736–745. https://doi.org/10.1073/pnas.1222809110
Arnold SJ, Dugger BN, Beach TG (2013) TDP-43 deposition in prospectively followed, cognitively normal elderly individuals: correlation with argyrophilic grains but not other concomitant pathologies. Acta Neuropathol 126:51–57. https://doi.org/10.1007/s00401-013-1110-0
Aulas A, Vande Velde C (2015) Alterations in stress granule dynamics driven by TDP-43 and FUS: a link to pathological inclusions in ALS? Front Cell Neurosci 9:423. https://doi.org/10.3389/fncel.2015.00423
Austin JA, Wright GS, Watanabe S, Grossmann JG, Antonyuk SV, Yamanaka K et al (2014) Disease causing mutants of TDP-43 nucleic acid binding domains are resistant to aggregation and have increased stability and half-life. Proc Natl Acad Sci USA 111:4309–4314. https://doi.org/10.1073/pnas.1317317111
Ayala YM, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D’Ambrogio A et al (2011) TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 30:277–288. https://doi.org/10.1038/emboj.2010.310
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C et al (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919. https://doi.org/10.1038/nature05016
Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K et al (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137:2329–2345. https://doi.org/10.1093/brain/awu138
Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM et al (2014) Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol 10:677–685. https://doi.org/10.1038/nchembio.1563
Bathgate D, Snowden JS, Varma A, Blackshaw A, Neary D (2001) Behaviour in frontotemporal dementia, Alzheimer’s disease and vascular dementia. Acta Neurol Scand 103:367–378
Bede P, Bokde AL, Byrne S, Elamin M, McLaughlin RL, Kenna K et al (2013) Multiparametric MRI study of ALS stratified for the C9orf72 genotype. Neurology 81:361–369. https://doi.org/10.1212/WNL.0b013e31829c5eee
Bede P, Hardiman O (2014) Lessons of ALS imaging: pitfalls and future directions—a critical review. Neuroimage Clin 4:436–443. https://doi.org/10.1016/j.nicl.2014.02.011
Benussi A, Padovani A, Borroni B (2015) Phenotypic heterogeneity of monogenic frontotemporal dementia. Front Aging Neurosci 7:171. https://doi.org/10.3389/fnagi.2015.00171
Bigio EH, Wu JY, Deng HX, Bit-Ivan EN, Mao Q, Ganti R et al (2013) Inclusions in frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP) and amyotrophic lateral sclerosis (ALS), but not FTLD with FUS proteinopathy (FTLD-FUS), have properties of amyloid. Acta Neuropathol 125:463–465. https://doi.org/10.1007/s00401-013-1089-6
Boxer AL, Mackenzie IR, Boeve BF, Baker M, Seeley WW, Crook R et al (2011) Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry 82:196–203. https://doi.org/10.1136/jnnp.2009.204081
Braak H, Ludolph A, Thal DR, Del Tredici K (2010) Amyotrophic lateral sclerosis: dash-like accumulation of phosphorylated TDP-43 in somatodendritic and axonal compartments of somatomotor neurons of the lower brainstem and spinal cord. Acta Neuropathol 120:67–74. https://doi.org/10.1007/s00401-010-0683-0
Braak H, Ludolph AC, Neumann M, Ravits J, Del Tredici K (2017) Pathological TDP-43 changes in Betz cells differ from those in bulbar and spinal alpha-motoneurons in sporadic amyotrophic lateral sclerosis. Acta Neuropathol 133:79–90. https://doi.org/10.1007/s00401-016-1633-2
Brettschneider J, Del Tredici K, Irwin DJ, Grossman M, Robinson JL, Toledo JB et al (2014) Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol 127:423–439. https://doi.org/10.1007/s00401-013-1238-y
Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M et al (2013) Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74:20–38. https://doi.org/10.1002/ana.23937
Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron D (2000) El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 1:293–299
Brunello CA, Yan X, Huttunen HJ (2016) Internalized Tau sensitizes cells to stress by promoting formation and stability of stress granules. Sci Rep 6:30498. https://doi.org/10.1038/srep30498
Buchan JR, Muhlrad D, Parker R (2008) P bodies promote stress granule assembly in Saccharomyces cerevisiae. J Cell Biol 183:441–455. https://doi.org/10.1083/jcb.200807043
Buratti E, De Conti L, Stuani C, Romano M, Baralle M, Baralle F (2010) Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J 277:2268–2281. https://doi.org/10.1111/j.1742-4658.2010.07643.x
Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE (2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 20:1774–1784. https://doi.org/10.1093/emboj/20.7.1774
Burrell JR, Halliday GM, Kril JJ, Ittner LM, Gotz J, Kiernan MC et al (2016) The frontotemporal dementia-motor neuron disease continuum. Lancet 388:919–931. https://doi.org/10.1016/S0140-6736(16)00737-6
Burrell JR, Kiernan MC, Vucic S, Hodges JR (2011) Motor neuron dysfunction in frontotemporal dementia. Brain 134:2582–2594. https://doi.org/10.1093/brain/awr195
Bury JJ, Highley JR, Cooper-Knock J, Goodall EF, Higginbottom A, McDermott CJ et al (2016) Oligogenic inheritance of optineurin (OPTN) and C9ORF72 mutations in ALS highlights localisation of OPTN in the TDP-43-negative inclusions of C9ORF72-ALS. Neuropathology 36:125–134. https://doi.org/10.1111/neup.12240
Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ et al (2007) TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171:227–240. https://doi.org/10.2353/ajpath.2007.070182
Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F et al (2018) TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci 21:228–239. https://doi.org/10.1038/s41593-017-0047-3
Cistaro A, Pagani M, Montuschi A, Calvo A, Moglia C, Canosa A et al (2014) The metabolic signature of C9ORF72-related ALS: FDG PET comparison with nonmutated patients. Eur J Nucl Med Mol Imaging 41:844–852. https://doi.org/10.1007/s00259-013-2667-5
Clerc P, Lipnick S, Willett C (2016) A look into the future of ALS research. Drug Discov Today 21:939–949. https://doi.org/10.1016/j.drudis.2016.02.002
Collinge J, Sidle KC, Meads J, Ironside J, Hill AF (1996) Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 383:685–690. https://doi.org/10.1038/383685a0
Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I et al (2014) Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128:755–766. https://doi.org/10.1007/s00401-014-1349-0
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D et al (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924. https://doi.org/10.1038/nature05017
D’Alton S, Altshuler M, Cannon A, Dickson DW, Petrucelli L, Lewis J (2014) Divergent phenotypes in mutant TDP-43 transgenic mice highlight potential confounds in TDP-43 transgenic modeling. PLoS One 9:e86513. https://doi.org/10.1371/journal.pone.0086513
Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du Plessis D, Neary D et al (2007) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol 113:521–533. https://doi.org/10.1007/s00401-006-0189-y
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256. https://doi.org/10.1016/j.neuron.2011.09.011
Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N et al (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477:211–215. https://doi.org/10.1038/nature10353
Devenney E, Hornberger M, Irish M, Mioshi E, Burrell J, Tan R et al (2014) Frontotemporal dementia associated with the C9ORF72 mutation: a unique clinical profile. JAMA Neurol 71:331–339. https://doi.org/10.1001/jamaneurol.2013.6002
Dickson DW, Weller RO (eds) (2011) Neurodegeneration: the molecular pathology of dementia and movement disorders, 2nd edn. Wiley and Blackwell, London
Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R et al (2018) Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561:137–140. https://doi.org/10.1038/s41586-018-0454-y
Fang YS, Tsai KJ, Chang YJ, Kao P, Woods R, Kuo PH et al (2014) Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat Commun 5:4824. https://doi.org/10.1038/ncomms5824
Feiler MS, Strobel B, Freischmidt A, Helferich AM, Kappel J, Brewer BM et al (2015) TDP-43 is intercellularly transmitted across axon terminals. J Cell Biol 211:897–911. https://doi.org/10.1083/jcb.201504057
Fekete T, Zach N, Mujica-Parodi LR, Turner MR (2013) Multiple kernel learning captures a systems-level functional connectivity biomarker signature in amyotrophic lateral sclerosis. PLoS One 8:e85190. https://doi.org/10.1371/journal.pone.0085190
Foulds P, McAuley E, Gibbons L, Davidson Y, Pickering-Brown SM, Neary D et al (2008) TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer’s disease and frontotemporal lobar degeneration. Acta Neuropathol 116:141–146. https://doi.org/10.1007/s00401-008-0389-8
Foulds PG, Davidson Y, Mishra M, Hobson DJ, Humphreys KM, Taylor M et al (2009) Plasma phosphorylated-TDP-43 protein levels correlate with brain pathology in frontotemporal lobar degeneration. Acta Neuropathol 118:647–658. https://doi.org/10.1007/s00401-009-0594-0
Fratta P, Sivakumar P, Humphrey J, Lo K, Ricketts T, Oliveira H et al (2018) Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J. https://doi.org/10.15252/embj.201798684
Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K et al (2015) Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 18:631–636. https://doi.org/10.1038/nn.4000
Fujishiro H, Uchikado H, Arai T, Hasegawa M, Akiyama H, Yokota O et al (2009) Accumulation of phosphorylated TDP-43 in brains of patients with argyrophilic grain disease. Acta Neuropathol 117:151–158. https://doi.org/10.1007/s00401-008-0463-2
Gasset-Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L et al (2019) Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102(339–357):e337. https://doi.org/10.1016/j.neuron.2019.02.038
Gautam M, Jara JH, Kocak N, Rylaarsdam LE, Kim KD et al (2019) Mitochondria, ER, and nuclear membrane defects reveal early mechanisms for upper motor neuron vulnerability with respect to TDP-43 pathology. Acta Neuropathol 137:47–69. https://doi.org/10.1007/s00401-018-1934-8
Geser F, Brandmeir NJ, Kwong LK, Martinez-Lage M, Elman L, McCluskey L et al (2008) Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol 65:636–641. https://doi.org/10.1001/archneur.65.5.636
Geser F, Martinez-Lage M, Kwong LK, Lee VM, Trojanowski JQ (2009) Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurol 256:1205–1214. https://doi.org/10.1007/s00415-009-5069-7
Goossens J, Vanmechelen E, Trojanowski JQ, Lee VM, Van Broeckhoven C, van der Zee J et al (2015) TDP-43 as a possible biomarker for frontotemporal lobar degeneration: a systematic review of existing antibodies. Acta Neuropathol Commun 3:15. https://doi.org/10.1186/s40478-015-0195-1
Gorno-Tempini ML, Dronkers NF, Rankin KP, Ogar JM, Phengrasamy L, Rosen HJ et al (2004) Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol 55:335–346. https://doi.org/10.1002/ana.10825
Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF et al (2011) Classification of primary progressive aphasia and its variants. Neurology 76:1006–1014. https://doi.org/10.1212/WNL.0b013e31821103e6
Grosskreutz J, Van Den Bosch L, Keller BU (2010) Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47:165–174. https://doi.org/10.1016/j.ceca.2009.12.002
Hardy J, Rogaeva E (2014) Motor neuron disease and frontotemporal dementia: sometimes related, sometimes not. Exp Neurol 262(Pt B):75–83. https://doi.org/10.1016/j.expneurol.2013.11.006
Hasegawa M, Arai T, Akiyama H, Nonaka T, Mori H, Hashimoto T et al (2007) TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. Brain 130:1386–1394. https://doi.org/10.1093/brain/awm065
Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y et al (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64:60–70. https://doi.org/10.1002/ana.21425
Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y et al (2008) Significance of the TDP-43 deposition in FTLD-U and ALS. Rinsho Shinkeigaku 48:994–997
Hasegawa M, Nonaka T, Masuda-Suzukake M (2017) Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders. Pharmacol Ther 172:22–33. https://doi.org/10.1016/j.pharmthera.2016.11.010
Hatanpaa KJ, Bigio EH, Cairns NJ, Womack KB, Weintraub S, Morris JC et al (2008) TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a midwest-southwest consortium for FTLD study. J Neuropathol Exp Neurol 67:271–279. https://doi.org/10.1097/NEN.0b013e31816a12a6
Homan P, Levy I, Feltham E, Gordon C, Hu J, Li J et al (2019) Neural computations of threat in the aftermath of combat trauma. Nat Neurosci 22:470–476. https://doi.org/10.1038/s41593-018-0315-x
Huey ED, Goveia EN, Paviol S, Pardini M, Krueger F, Zamboni G et al (2009) Executive dysfunction in frontotemporal dementia and corticobasal syndrome. Neurology 72:453–459. https://doi.org/10.1212/01.wnl.0000341781.39164.26
Iguchi Y, Eid L, Parent M, Soucy G, Bareil C, Riku Y et al (2016) Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 139:3187–3201. https://doi.org/10.1093/brain/aww237
Irwin DJ, McMillan CT, Brettschneider J, Libon DJ, Powers J et al (2013) Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 84:163–169. https://doi.org/10.1136/jnnp-2012-303507
Jackson M, Lennox G, Lowe J (1996) Motor neurone disease-inclusion dementia. Neurodegeneration 5:339–350
Ji AL, Zhang X, Chen WW, Huang WJ (2017) Genetics insight into the amyotrophic lateral sclerosis/frontotemporal dementia spectrum. J Med Genet 54:145–154. https://doi.org/10.1136/jmedgenet-2016-104271
Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM et al (2011) Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 122:137–153. https://doi.org/10.1007/s00401-011-0839-6
Josephs KA, Mackenzie I, Frosch MP, Bigio EH, Neumann M, Arai T et al (2019) LATE to the PART-y. Brain. https://doi.org/10.1093/brain/awz224
Josephs KA, Murray ME, Tosakulwong N, Weigand SD, Knopman DS, Petersen RC et al (2019) Brain atrophy in primary age-related tauopathy is linked to transactive response DNA-binding protein of 43 kDa. Alzheimers Dement 15:799–806. https://doi.org/10.1016/j.jalz.2019.03.003
Josephs KA, Murray ME, Tosakulwong N, Weigand SD, Serie AM, Perkerson RB et al (2019) Pathological, imaging and genetic characteristics support the existence of distinct TDP-43 types in non-FTLD brains. Acta Neuropathol 137:227–238. https://doi.org/10.1007/s00401-018-1951-7
Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L et al (2016) Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol 131:571–585. https://doi.org/10.1007/s00401-016-1537-1
Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM et al (2014) TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 127:811–824. https://doi.org/10.1007/s00401-014-1269-z
Junttila A, Kuvaja M, Hartikainen P, Siloaho M, Helisalmi S, Moilanen V et al (2016) Cerebrospinal fluid TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis patients with and without the C9ORF72 hexanucleotide expansion. Dement Geriatr Cogn Dis Extra 6:142–149. https://doi.org/10.1159/000444788
Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574. https://doi.org/10.1038/ng.132
Kametani F, Obi T, Shishido T, Akatsu H, Murayama S, Saito Y et al (2016) Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci Rep 6:23281. https://doi.org/10.1038/srep23281
Kapeli K, Martinez FJ, Yeo GW (2017) Genetic mutations in RNA-binding proteins and their roles in ALS. Hum Genet 136:1193–1214. https://doi.org/10.1007/s00439-017-1830-7
Kim EJ, Sidhu M, Gaus SE, Huang EJ, Hof PR, Miller BL et al (2016) Selective frontoinsular von Economo neuron and fork cell loss in early behavioral variant frontotemporal dementia. Cereb Cortex 26:1843. https://doi.org/10.1093/cercor/bhw012
Kovacs GG (2016) Molecular pathological classification of neurodegenerative diseases: turning towards precision medicine. Int J Mol Sci. https://doi.org/10.3390/ijms17020189
Kovacs GG, Botond G, Budka H (2010) Protein coding of neurodegenerative dementias: the neuropathological basis of biomarker diagnostics. Acta Neuropathol 119:389–408. https://doi.org/10.1007/s00401-010-0658-1
Kovacs GG, Murrell JR, Horvath S, Haraszti L, Majtenyi K, Molnar MJ et al (2009) TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord 24:1843–1847. https://doi.org/10.1002/mds.22697
Kuiperij HB, Abdo WF, van Engelen BG, Schelhaas HJ, Verbeek MM (2010) TDP-43 plasma levels do not differentiate sporadic inclusion body myositis from other inflammatory myopathies. Acta Neuropathol 120:825–826. https://doi.org/10.1007/s00401-010-0769-8
Kuo PH, Doudeva LG, Wang YT, Shen CK, Yuan HS (2009) Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res 37:1799–1808. https://doi.org/10.1093/nar/gkp013
Lagier-Tourenne C, Polymenidou M, Cleveland DW (2010) TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet 19:R46–64. https://doi.org/10.1093/hmg/ddq137
Lee EB, Porta S, Michael Baer G, Xu Y, Suh E, Kwong LK et al (2017) Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol 134:65–78. https://doi.org/10.1007/s00401-017-1679-9
Lee SE, Khazenzon AM, Trujillo AJ, Guo CC, Yokoyama JS, Sha SJ et al (2014) Altered network connectivity in frontotemporal dementia with C9orf72 hexanucleotide repeat expansion. Brain 137:3047–3060. https://doi.org/10.1093/brain/awu248
Lillo P, Garcin B, Hornberger M, Bak TH, Hodges JR (2010) Neurobehavioral features in frontotemporal dementia with amyotrophic lateral sclerosis. Arch Neurol 67:826–830. https://doi.org/10.1001/archneurol.2010.146
Lomen-Hoerth C, Anderson T, Miller B (2002) The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59:1077–1079
Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ et al (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61:427–434. https://doi.org/10.1002/ana.21147
Mackenzie IR, Frick P, Neumann M (2014) The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol 127:347–357. https://doi.org/10.1007/s00401-013-1232-4
Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E et al (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122:111–113. https://doi.org/10.1007/s00401-011-0845-8
Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J et al (2009) Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol 117:15–18. https://doi.org/10.1007/s00401-008-0460-5
Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C et al (2017) TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95(808–816):e809. https://doi.org/10.1016/j.neuron.2017.07.025
Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T et al (2012) Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain 135:736–750. https://doi.org/10.1093/brain/awr361
Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis-Marks MR, Needham PG et al (2019) RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 102(321–338):e328. https://doi.org/10.1016/j.neuron.2019.01.048
McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I et al (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131:75–86. https://doi.org/10.1007/s00401-015-1515-z
McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW et al (2010) TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 69:918–929. https://doi.org/10.1097/NEN.0b013e3181ee7d85
McKee AC, Robinson ME (2014) Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement 10:S242–253. https://doi.org/10.1016/j.jalz.2014.04.003
McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136:43–64. https://doi.org/10.1093/brain/aws307
Meyer H, Weihl CC (2014) The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci 127:3877–3883. https://doi.org/10.1242/jcs.093831
Nana AL, Sidhu M, Gaus SE, Hwang JL, Li L, Park Y et al (2019) Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol 137:27–46. https://doi.org/10.1007/s00401-018-1942-8
Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K et al (2019) Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142:1503–1527. https://doi.org/10.1093/brain/awz099
Nelson PT, Schmitt FA, Lin Y, Abner EL, Jicha GA, Patel E et al (2011) Hippocampal sclerosis in advanced age: clinical and pathological features. Brain 134:1506–1518. https://doi.org/10.1093/brain/awr053
Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM (2007) TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol 64:1388–1394. https://doi.org/10.1001/archneur.64.10.1388
Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA et al (2007) TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol 66:177–183. https://doi.org/10.1097/01.jnen.0000248554.45456.58
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. https://doi.org/10.1126/science.1134108
Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T et al (2013) Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep 4:124–134. https://doi.org/10.1016/j.celrep.2013.06.007
Okamoto K, Fujita Y, Mizuno Y (2010) Pathology of protein synthesis and degradation systems in ALS. Neuropathology 30:189–193. https://doi.org/10.1111/j.1440-1789.2009.01088.x
Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB (1995) Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69:3584–3596
Parchi P, Capellari S, Chen SG, Petersen RB, Gambetti P, Kopp N et al (1997) Typing prion isoforms. Nature 386:232–234. https://doi.org/10.1038/386232a0
Peters F, Perani D, Herholz K, Holthoff V, Beuthien-Baumann B, Sorbi S et al (2006) Orbitofrontal dysfunction related to both apathy and disinhibition in frontotemporal dementia. Dement Geriatr Cogn Disord 21:373–379. https://doi.org/10.1159/000091898
Porta S, Xu Y, Restrepo CR, Kwong LK, Zhang B, Brown HJ et al (2018) Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat Commun 9:4220. https://doi.org/10.1038/s41467-018-06548-9
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J et al (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134:2456–2477. https://doi.org/10.1093/brain/awr179
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268. https://doi.org/10.1016/j.neuron.2011.09.010
Riggs JE (2001) The latency between traumatic axonal injury and the onset of amyotrophic lateral sclerosis in young adult men. Mil Med 166:731–732
Riggs JE, Hobbs GR (2003) Motor axonal injury and amyotrophic lateral sclerosis: risk assessment using a reverse probability analysis technique. Mil Med 168:143–145
Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE (2005) Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65:586–590. https://doi.org/10.1212/01.wnl.0000172911.39167.b6
Rosen DR (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364:362. https://doi.org/10.1038/364362c0
Scholl M, Damian A, Engler H (2014) Fluorodeoxyglucose PET in neurology and psychiatry. PET Clin 9(371–390):v. https://doi.org/10.1016/j.cpet.2014.07.005
Schwab C, Arai T, Hasegawa M, Akiyama H, Yu S, McGeer PL (2009) TDP-43 pathology in familial British dementia. Acta Neuropathol 118:303–311. https://doi.org/10.1007/s00401-009-0514-3
Seelaar H, Rohrer JD, Pijnenburg YA, Fox NC, van Swieten JC (2011) Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry 82:476–486. https://doi.org/10.1136/jnnp.2010.212225
Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD (2009) Neurodegenerative diseases target large-scale human brain networks. Neuron 62:42–52. https://doi.org/10.1016/j.neuron.2009.03.024
Sha SJ, Takada LT, Rankin KP, Yokoyama JS, Rutherford NJ, Fong JC et al (2012) Frontotemporal dementia due to C9ORF72 mutations: clinical and imaging features. Neurology 79:1002–1011. https://doi.org/10.1212/WNL.0b013e318268452e
Shaw PJ, Eggett CJ (2000) Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J Neurol 247(Suppl 1):I17–27
Shimonaka S, Nonaka T, Suzuki G, Hisanaga S, Hasegawa M (2016) Templated aggregation of TAR DNA-binding protein of 43 kDa (TDP-43) by seeding with TDP-43 peptide fibrils. J Biol Chem 291:8896–8907. https://doi.org/10.1074/jbc.M115.713552
Shimozawa A, Ono M, Takahara D, Tarutani A, Imura S, Masuda-Suzukake M et al (2017) Propagation of pathological alpha-synuclein in marmoset brain. Acta Neuropathol Commun 5:12. https://doi.org/10.1186/s40478-017-0413-0
Snowden J, Neary D, Mann D (2007) Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol 114:31–38. https://doi.org/10.1007/s00401-007-0236-3
Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM et al (2012) Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain 135:693–708. https://doi.org/10.1093/brain/awr355
Spiller KJ, Restrepo CR, Khan T, Dominique MA, Fang TC, Canter RG et al (2018) Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat Neurosci 21:329–340. https://doi.org/10.1038/s41593-018-0083-7
Steinacker P, Barschke P, Otto M (2018) Biomarkers for diseases with TDP-43 pathology. Mol Cell Neurosci. https://doi.org/10.1016/j.mcn.2018.10.003
Steinacker P, Hendrich C, Sperfeld AD, Jesse S, von Arnim CA, Lehnert S et al (2008) TDP-43 in cerebrospinal fluid of patients with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol 65:1481–1487. https://doi.org/10.1001/archneur.65.11.1481
Talbot K (2014) Amyotrophic lateral sclerosis: cell vulnerability or system vulnerability? J Anat 224:45–51. https://doi.org/10.1111/joa.12107
Tan CF, Eguchi H, Tagawa A, Onodera O, Iwasaki T, Tsujino A et al (2007) TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol 113:535–542. https://doi.org/10.1007/s00401-007-0206-9
Tanaka Y, Chambers JK, Matsuwaki T, Yamanouchi K, Nishihara M (2014) Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol Commun 2:78. https://doi.org/10.1186/s40478-014-0078-x
Tanaka Y, Suzuki G, Matsuwaki T, Hosokawa M, Serrano G, Beach TG et al (2017) Progranulin regulates lysosomal function and biogenesis through acidification of lysosomes. Hum Mol Genet 26:969–988. https://doi.org/10.1093/hmg/ddx011
Thomsen GM, Vit JP, Lamb A, Gowing G, Shelest O, Alkaslasi M et al (2015) Acute traumatic brain injury does not exacerbate amyotrophic lateral sclerosis in the SOD1 (G93A) rat model. eNeuro. https://doi.org/10.1523/eneuro.0059-14.2015
Tsuji H, Arai T, Kametani F, Nonaka T, Yamashita M, Suzukake M et al (2012) Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain 135:3380–3391. https://doi.org/10.1093/brain/aws230
van der Zee J, Van Langenhove T, Kovacs GG, Dillen L, Deschamps W, Engelborghs S et al (2014) Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol 128:397–410. https://doi.org/10.1007/s00401-014-1298-7
Van Mossevelde S, Engelborghs S, van der Zee J, Van Broeckhoven C (2018) Genotype-phenotype links in frontotemporal lobar degeneration. Nat Rev Neurol 14:363–378. https://doi.org/10.1038/s41582-018-0009-8
Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PEA, Cook C, Lummertz da Rocha E et al (2016) Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep 15:1455–1466. https://doi.org/10.1016/j.celrep.2016.04.045
Volkening K, Leystra-Lantz C, Yang W, Jaffee H, Strong MJ (2009) Tar DNA binding protein of 43 kDa (TDP-43), 14–3–3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res 1305:168–182. https://doi.org/10.1016/j.brainres.2009.09.105
Walker AK, Daniels CM, Goldman JE, Trojanowski JQ, Lee VM, Messing A (2014) Astrocytic TDP-43 pathology in Alexander disease. J Neurosci 34:6448–6458. https://doi.org/10.1523/JNEUROSCI.0248-14.2014
Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D et al (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 36:377–381. https://doi.org/10.1038/ng1332
White MA, Kim E, Duffy A, Adalbert R, Phillips BU, Peters OM et al (2018) TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat Neurosci 21:552–563. https://doi.org/10.1038/s41593-018-0113-5
Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, DeJesus-Hernandez M et al (2012) Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 135:794–806. https://doi.org/10.1093/brain/aws001
Wiesner D, Tar L, Linkus B, Chandrasekar A, Olde Heuvel F, Dupuis L et al (2018) Reversible induction of TDP-43 granules in cortical neurons after traumatic injury. Exp Neurol 299:15–25. https://doi.org/10.1016/j.expneurol.2017.09.011
Wightman G, Anderson VE, Martin J, Swash M, Anderton BH, Neary D et al (1992) Hippocampal and neocortical ubiquitin-immunoreactive inclusions in amyotrophic lateral sclerosis with dementia. Neurosci Lett 139:269–274
Williams KL, Topp S, Yang S, Smith B, Fifita JA, Warraich ST et al (2016) CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun 7:11253. https://doi.org/10.1038/ncomms11253
Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM (2008) Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem 283:13302–13309. https://doi.org/10.1074/jbc.M800342200
Wolozin B (2019) The evolution of phase-separated TDP-43 in stress. Neuron 102:265–267. https://doi.org/10.1016/j.neuron.2019.03.041
Woolley JD, Gorno-Tempini ML, Seeley WW, Rankin K, Lee SS, Matthews BR et al (2007) Binge eating is associated with right orbitofrontal-insular-striatal atrophy in frontotemporal dementia. Neurology 69:1424–1433. https://doi.org/10.1212/01.wnl.0000277461.06713.23
Yu L, De Jager PL, Yang J, Trojanowski JQ, Bennett DA, Schneider JA (2015) The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology 84:927–934. https://doi.org/10.1212/WNL.0000000000001313
Zamboni G, Huey ED, Krueger F, Nichelli PF, Grafman J (2008) Apathy and disinhibition in frontotemporal dementia: insights into their neural correlates. Neurology 71:736–742. https://doi.org/10.1212/01.wnl.0000324920.96835.95
Zetterberg H, van Swieten JC, Boxer AL, Rohrer JD (2019) Review: fluid biomarkers for frontotemporal dementias. Neuropathol Appl Neurobiol 45:81–87. https://doi.org/10.1111/nan.12530
Zhou J, Gennatas ED, Kramer JH, Miller BL, Seeley WW (2012) Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron 73:1216–1227. https://doi.org/10.1016/j.neuron.2012.03.004
Zhou J, Greicius MD, Gennatas ED, Growdon ME, Jang JY, Rabinovici GD et al (2010) Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer’s disease. Brain 133:1352–1367. https://doi.org/10.1093/brain/awq075
Acknowledgements
We would like to thank Dr. Takashi Nonaka (Dementia Research Project, Tokyo Metropolitan Institute of Medical Science), Prof. Shigeo Murayama (Department of Neuropathology, Tokyo Metropolitan Geriatric Hospital and Institute) and Prof. Emer. Kinuko Suzuki (Department of Neurology and Psychiatry, University of North Carolina at Chapel Hill) for helpful advice and support. Also, we thank Ms Hiromi Kondo, Chie Haga and Yoko Shimomura (Dementia Research Project, Tokyo Metropolitan Institute of Medical Science) for their technical assistance. This work was supported by The Naito Foundation (to I.K.) and Ministry of Education, Culture, Sports, Science, and Technology Grants-in-Aid for Scientific Research Grants JP26117005 (to M.H.), Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research Grant JP23228004 (to M.H.), and a grant-in-aid for research on Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) from the Japan Agency for Medical Research and Development (AMED) JP14533254 (to M.H.).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kawakami, I., Arai, T. & Hasegawa, M. The basis of clinicopathological heterogeneity in TDP-43 proteinopathy. Acta Neuropathol 138, 751–770 (2019). https://doi.org/10.1007/s00401-019-02077-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-019-02077-x