Article Text

Abstract
Background Schaaf-Yang syndrome (SYS) is caused by truncating mutations in MAGEL2, mapping to the Prader-Willi region (15q11-q13), with an observed phenotype partially overlapping that of Prader-Willi syndrome. MAGEL2 plays a role in retrograde transport and protein recycling regulation. Our aim is to contribute to the characterisation of SYS pathophysiology at clinical, genetic and molecular levels.
Methods We performed an extensive phenotypic and mutational revision of previously reported patients with SYS. We analysed the secretion levels of amyloid-β 1–40 peptide (Aβ1-40) and performed targeted metabolomic and transcriptomic profiles in fibroblasts of patients with SYS (n=7) compared with controls (n=11). We also transfected cell lines with vectors encoding wild-type (WT) or mutated MAGEL2 to assess stability and subcellular localisation of the truncated protein.
Results Functional studies show significantly decreased levels of secreted Aβ1-40 and intracellular glutamine in SYS fibroblasts compared with WT. We also identified 132 differentially expressed genes, including non-coding RNAs (ncRNAs) such as HOTAIR, and many of them related to developmental processes and mitotic mechanisms. The truncated form of MAGEL2 displayed a stability similar to the WT but it was significantly switched to the nucleus, compared with a mainly cytoplasmic distribution of the WT MAGEL2. Based on the updated knowledge, we offer guidelines for the clinical management of patients with SYS.
Conclusion A truncated MAGEL2 protein is stable and localises mainly in the nucleus, where it might exert a pathogenic neomorphic effect. Aβ1-40 secretion levels and HOTAIR mRNA levels might be promising biomarkers for SYS. Our findings may improve SYS understanding and clinical management.
- clinical genetics
- gene expression
- central nervous system diseases
- disease management
- nervous system diseases
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- clinical genetics
- gene expression
- central nervous system diseases
- disease management
- nervous system diseases
WHAT IS ALREADY KNOWN ON THIS TOPIC
MAGEL2-truncating mutations cause Schaaf-Yang syndrome (SYS), but the functional effects of the truncated MAGEL2 protein have been poorly defined.
WHAT THIS STUDY ADDS
By expressing truncated MAGEL2 in cell lines, we now know that a truncated version of the protein is retained in the nucleus, thus exerting a novel behaviour in addition to the loss of some of its main functions. Patients’ fibroblasts show reduced levels of excreted amyloid-β 1–40 and intracellular glutamine as well as an altered transcriptomic profile, including overexpression of the major regulator HOTAIR.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Based on a comprehensive review of genetic and clinical aspects of all reported cases, families and physicians will benefit from the Clinical Management Recommendations that we provide here.
Introduction
In 2013, truncating mutations in MAGEL2 (OMIM 605283) were associated with a new clinical entity,1 first described as a Prader-Willi-like syndrome and currently named as Schaaf-Yang syndrome (SYS, OMIM 615547). MAGEL2 is one of the five maternally imprinted protein-coding genes contained in the Prader-Willi region (15q11-q13). Lack of expression of the paternal alleles in this region causes Prader-Willi syndrome (PWS; OMIM 176270). In contrast, non-sense or frameshift mutations in the paternal allele of MAGEL2 are predicted to encode a truncated protein lacking the MAGE Homology Domain (MHD) and have been associated with SYS.1 Since then, over a hundred patients with SYS have been reported and phenotypically described.1–28 Patients with SYS and PWS show overlapping clinical phenotypes, including neonatal hypotonia, intellectual disability (ID), developmental delay (DD), early feeding difficulties, endocrinological disturbances (hypogonadism and other hormonal imbalances) and sleep disorders. However, some of the clinical criteria for PWS diagnosis, such as hypopigmentation, characteristic facial dysmorphisms, small hand and feet, hyperphagia, obesity and obsessive-compulsive behaviours, are frequently absent in patients with SYS, who, on the other hand, present more frequently with severe ID, autism spectrum disorder (ASD) behaviours and joint contractures.29–31 Some truncating variants in MAGEL2 have also been associated with Chitayat-Hall syndrome (CHS).32 However, a systematic review of all patients with SYS and CHS showed that there is no discernible genetic or clinical difference between both syndromes, and the latter has been renamed as SYS in OMIM.17 In contrast, two particular MAGEL2 truncating mutations have been recurrently identified in patients affected by lethal arthrogryposis multiplex congenita (AMC), a much more severe phenotype, distinct from SYS.4 10 27 33 34 All in all, there is no specific constellation of symptoms pathognomonic or specific for any of these clinical syndromes; furthermore, they probably conform to a clinical continuum, therefore denoting the need to address clinical denomination according to molecular findings.
MAGEL2 shows a wider expression in human fetal tissues than in adult tissues, where it is predominantly present in brain (according to GTEx35). In adult mice, it also becomes mostly restricted to the central nervous system, specifically to the amygdala and the hypothalamus, and predominantly in the suprachiasmatic, the paraventricular and the supraoptic nuclei.36–38 It is a single-exon gene that encodes one of the largest proteins of the type II MAGE protein family consisting of 1249 amino acids. At a structural level, the N-terminal region of MAGEL2 contains a proline-rich domain, whose function remains unclear.39 At the C-terminus, from amino acids 1027 to 1195, there is the MHD, a highly conserved 170-amino acid sequence present in both type I and type II MAGEs, crucial for protein–protein interaction.40 Through it, MAGEL2 recognises and binds the coiled-coil domain of the E3 ubiquitin ligase TRIM27. The MHD is also crucial for binding VPS35, a subunit of the retromer cargo-selective complex. MAGEL2, TRIM27 and USP7 form the MUST complex, which is recruited to endosomes through direct binding of MAGEL2 to VPS35 and plays a role in retrograde endosomal transport.41 42 These specialised endosomes participate in endosomal export pathways that deliver membrane protein cargoes either to the trans-Golgi network through retrograde pathways or to the plasma membrane through recycling pathways.43
A dysfunction of the retrograde transport could be disturbing for many cellular processes. Loss of MAGEL2 expression causes a reduction in secretory granules protein levels due to impaired endosomal protein trafficking and subsequent lysosomal degradation, resulting in a reduction of circulating bioactive hypothalamic hormones.36 A well-coordinated trafficking network is also key for the correct regulation of amyloid precursor protein (APP) cleavage.44 APP family members are relevant for neuronal differentiation and migration during cortical development45 46 and proper neuromuscular junction formation and neurotransmission.47 Many studies support a model where retromer deficiency leads to increased APP cleavage, Aβ peptide production and exocytosis.48–50 In addition, protein levels of the glucose transporter GLUT1 in the cell membrane are reduced after VPS35 and SNX27 inhibition, showing that they are also tightly regulated by the retromer.51
Here, we have performed an extensive literature revision and based on it, we have expanded the clinical and genetic delineation of SYS and developed a standardised set of guidelines for its clinical management. We also contribute to the knowledge of the cellular phenotype by assessing the effect of a recurrent truncating variant on MAGEL2 protein stability and subcellular localisation using heterologous expression vectors. Finally, we have performed a transcriptomic and metabolomic characterisation of fibroblasts derived from patients with SYS and interpreted the results in the context of molecular and clinical findings.
Results
Clinical management of patients with SYS requires a coordinated multidisciplinary approach
We performed a systematic revision of all the published patients with SYS to date, who were all carriers of MAGEL2 mutations (table 1 and online supplemental table 1), carefully inspecting both the molecular and phenotypic data, with the aim to propose a set of guidelines for SYS clinical management.
Supplemental material
Clinical overview of patients with MAGEL2-related disorders
The literature-based recommendations have been divided into two life periods: the perinatal period (first 28 days of life) and infancy/adolescence. They include the most relevant medical problems associated with each period and the specific concerns or interventions advisable for each area. A schematic version of the different clinical areas and tests included in the guidelines are represented in figure 1, and a detailed, printable version is supplied as online supplemental table 2 (both in Spanish as online supplemental figure 1 and online supplemental table 3).
Supplemental material
Supplemental material
Supplemental material
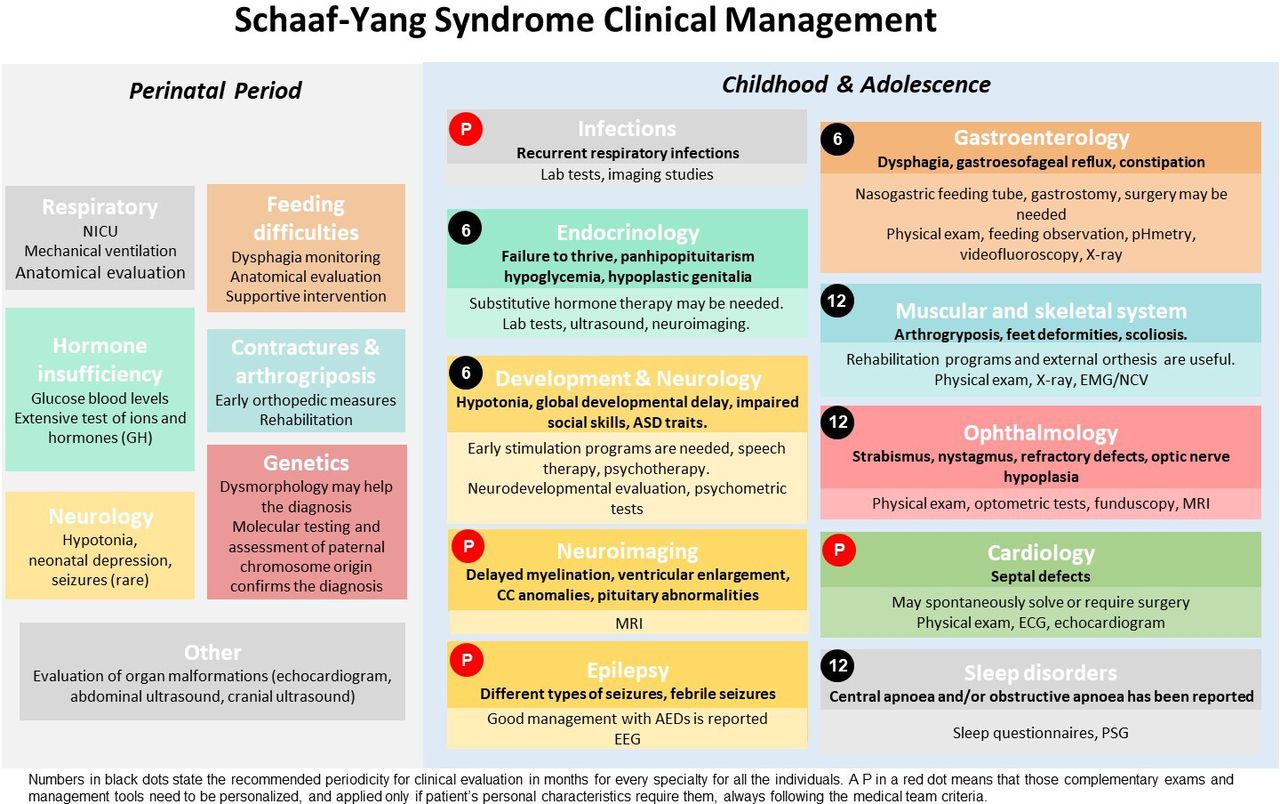
Schematic guidelines for Schaaf-Yang syndrome clinical management.AEDs, anti epileptic drugs; ASD, autism spectrum disorder; CC, corpus callosum; EEG, electroencephalogram; EMG/NCV, electromyogram/nerve conduction velocity; GH, growth hormone; NICU, neonatal intensive care unit; PSG, polysomnography. Perinatal period: Boxes include the medical area of disease. Medical problems and management requirements are detailed below. Childhood and adolescence: Boxes include medical area of disease. Medical problems and diagnosis are detailed below in bold. Management recommendations and complementary examinations are below, not in bold letters with lighter colours at the bottom. Numbers in black dots state the recommended periodicity for clinical evaluation in months for every specialty for all the individuals. In contrast, a P in a red dot means that those complementary examinations and management tools need to be personalised, and applied only if patient’s personal characteristics require them, always following the medical team criteria. For instance, MRI and cardiology are recommended at diagnosis/birth for all patients, but follow-up depends on comorbidities. Please see online supplemental table 2 for more details. A Spanish version of this figure is included as online supplemental figure 1.
In most instances, pregnancy was uneventful (only polyhydramnios has been reported in some cases), although there is a high rate of caesarean sections (over 50% of the patients where delivery information is available). Clinical symptoms appear early in life showing a complex phenotype that includes neuromuscular symptoms, respiratory and endocrinological problems, feeding difficulties and dysmorphic traits. Other clinical issues can appear later and may affect almost any organ or system, requiring a coordinated multidisciplinary approach.
SYS variants are mostly truncating and located in the C-terminal domain
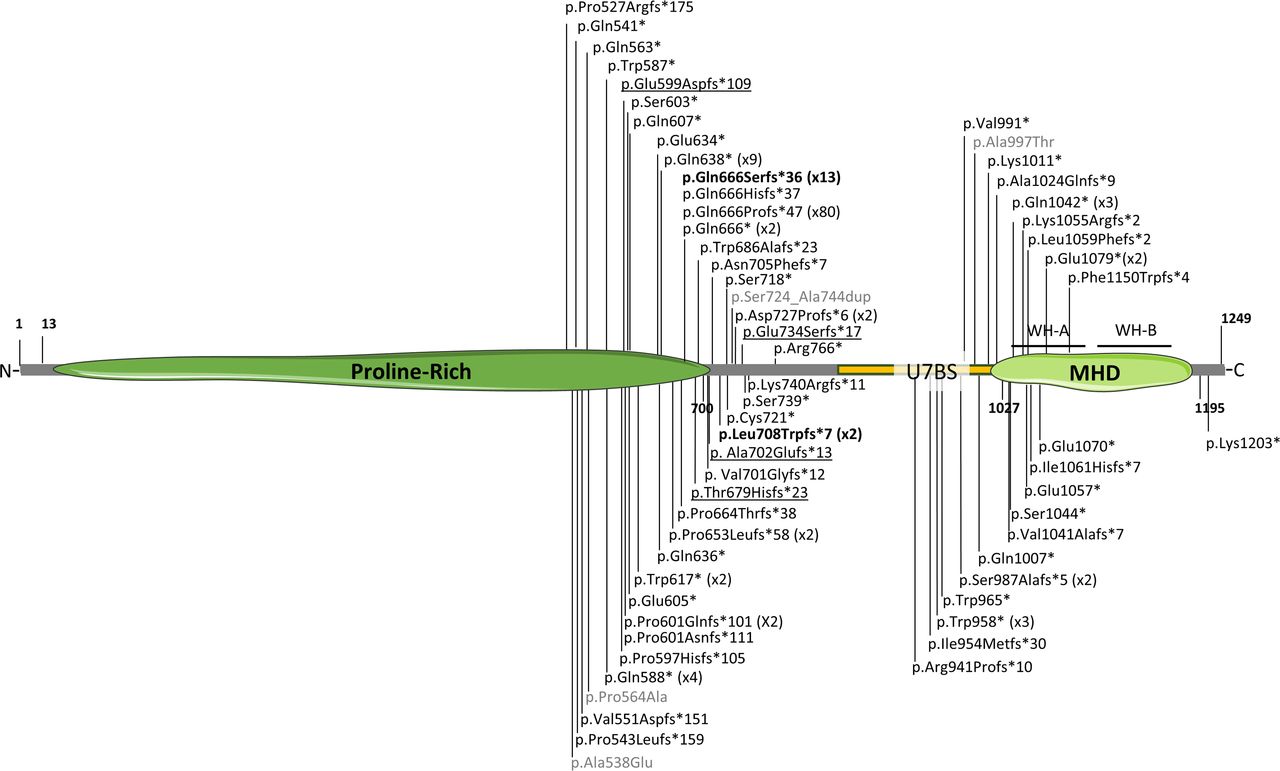
To date, 61 different variants have been associated with SYS or AMC (figure 2). Mutation p.(Gln666Profs*47), present in 80 individuals, is considered a recurrent mutational hotspot. These variants are mostly truncating and predominantly located in the C-terminal region of the protein, leading to a partial or a total lack of the MHD domain and compromising the functions that MAGEL2 carries out through this domain. Three different missense variants and a small in-frame duplication have also been associated with SYS phenotypes (in grey in figure 2). These atypical variants are not predicted to encode a truncated form of the protein, and there are no functional studies to support their pathogenicity. Thus, their clinical implication remains to be proven.

Schematic representation of MAGEL2 disease-associated variants. MHD, MAGE Homology Domain. The number in brackets indicates the number of individuals carrying recurrent mutations. In bold, mutations p.Gln666Serfs*36 (x13) and p.Leu708Trpfs*7 (x2) associated with arthrogryposis multiplex congenita. In grey, atypical, non-truncating variants. Previously misreported variants c.1797_1820del (p.Glu599Aspfs*109 (c.1797_1810del)), c.224delC (p.Thyr679Hisfs*23 (c.2034delC)), c.2015delC (p.Ala702Glufs*13 (c.2105delC)) and c.390delA (p.Glu734Serfs*17 (c.2199delA))10 are underlined. All variants are referenced to the MAGEL2 hg38 transcript NM_019066.5. The complete list of variants, their cDNA annotation and reference are collected in online supplemental table 4. Image created with Servier Medical Art (smart.servier.com).
Supplemental material
Fibroblasts from patients with SYS show altered gene expression patterns
To better understand the effect of MAGEL2 truncating mutations on gene expression patterns, we performed an mRNA whole transcriptome analysis (mRNASeq) on fibroblasts from six healthy donors and three SYS subjects: a girl carrying p.(Gln638*) and a boy and an unrelated girl, both carrying p.(Gln666Profs*47). Using the ExpHunter Suite, we identified 132 differentially expressed genes, 76 upregulated and 56 downregulated (online supplemental table 4 and online supplemental figure 2A,B). The top 10 upregulated and downregulated genes, showing the most significant changes in the expression fold, are listed in table 2. Four genes were tested by quantitative PCR (qPCR) in fibroblasts from five patients with SYS and four control individuals, which confirmed significant upregulation of HOTAIR and PITX1 and downregulation of TBX5 (online supplemental figure 2C).
Top 10 upregulated and downregulated DEGs identified after mRNASeq analysis of skin fibroblasts in patients with SYS and controls
Enrichment analysis on the 132 identified DEGs highlighted a group of five genes related to ‘collagen formation’ and various mitosis-related REACTOME categories, such as ‘Resolution of Sister Chromatid Cohesion’ and ‘Mitotic Spindle Checkpoint’ (online supplemental figure 2D). Consistently, most of those genes were also present in the ‘Kinetochore’ and ‘Chromosome, centromeric region’ categories according to Gene Ontology cellular component enrichment (online supplemental table 5).
SYS fibroblasts show decreased Aβ1-40 peptide secretion levels
Given MAGEL2 function in the retromer, the truncation of MAGEL2 could affect APP cleavage and Aβ1-40 peptide production rates. Thus, levels of Aβ1-40 and Aβ1-42 peptides were measured by ELISA in the extracellular medium from SYS, PWS and control fibroblasts in the search for a potential biomarker of truncated MAGEL2 function. SYS fibroblasts showed significantly decreased extracellular levels of the Aβ1-40 processed peptide, both compared with PWS and control fibroblasts. No differences were observed in the PWS group compared with the control (figure 3A). Levels of Aβ1-42 peptide were extremely low and no differences were observed between conditions (data not shown).

Molecular and cellular biomarkers for Schaaf-Yang syndrome (SYS). (A) Amyloid-β 1–40 (Aβ1-40) peptide levels in control, Prader-Willi syndrome (PWS) and SYS fibroblasts’ extracellular medium. Data obtained from at least three independent experiments (SYS: n=5; PWS: n=9; control: n=5). (B) Glutamine levels in control, PWS and SYS fibroblasts (SYS: n=6; PWS: n=9; control: n=6). Values from two independent experiments have been normalised to the mean of the control group. Horizontal lines represent mean values and error bars represent the SD. Statistical analyses were performed using one-way analysis of variance and Tukey’s multiple comparisons test in GraphPad Prism. *p<0.001. (C) Colocalisation quantification using Mander’s coefficient between the haemagglutinin (HA) fluorescence signal and DAPI (4′,6-diamidino-2-phenylindole) in MAGEL2-wild-type (WT) and MAGEL2-Gln638* transfected cells normalised by the total HA intensity. n=118 from six independent experiments. *p<0.001. (D) Representative immunofluorescence images of SAOS-2 cells transfected with MAGEL2-WT and MAGEL2-Gln638* plasmids, stained with anti-HA (green, tagging MAGEL2) or DAPI (blue, cell nuclei). Scale bar represents 15 µm.
SYS fibroblasts show altered levels of organic acids and amino acids (metabolic profiling in fibroblasts)
Mass spectrometry analysis of intracellular metabolites from extracts of SYS (n=4), PWS (n=9) and control (n=5) fibroblasts showed a robust and significant decrease in glutamine levels for SYS fibroblasts (figure 3B), as well as a significant increase of suberic, sebacic, adipic and malic organic acids levels (online supplemental figure 3).
We investigated if any of the genes involved in these metabolites’ pathways were differentially expressed in the transcriptomic analysis (online supplemental table 4). The only differentially expressed gene involved in glutamine (GO:0006541) or organic acid (GO:0006082) metabolic processes was ME1 (ENSG00000065833), encoding the NADP-dependent malic enzyme protein, which was upregulated in SYS fibroblasts.
Truncated form of MAGEL2 remains stable in the cell
To evaluate the stability and recycling of a truncated form of MAGEL2, HEK293T cells were transfected with haemagglutinin (HA)-tagged expression vectors containing either the wild-type (WT) cDNA sequence of MAGEL2 (MAGEL2-WT) or the c.1912C>T; p.Gln638* (MAGEL2-Gln638*) mutation, which encodes a protein 611 amino acids shorter than the WT form and has been reported in nine patients (online supplemental table 1). Blocking of the proteasomal degradation pathway with MG132 (online supplemental figure 4A) or the lysosomal pathway with bafilomycin (online supplemental figure 4B) showed that MAGEL2 is mostly degraded via proteasome with no differences between WT and truncated MAGEL2 (online supplemental figure 4A,B). A time-course of cycloheximide treatment (2, 4, 6, 12 hours) showed also a similar stability and half-life for both MAGEL2 forms (online supplemental figure 4C).
Increased nuclear localisation of the truncated form of MAGEL2
To determine the subcellular localisation of the p.Gln638* truncated form of MAGEL2, HEK293T, SAOS-2 and HeLa cells were transfected with the expression vectors MAGEL2-WT or MAGEL2-Gln638*. Immunocytochemistry assays detecting HA showed that in SAOS-2 cells transfected with the MAGEL2-Gln638* construct and quantified by Mander’s coefficient there was a shift of the protein towards the nucleus, while the MAGEL2-WT form was mainly located in the cytoplasm (figure 3C,D). A similar localisation pattern was observed in HEK293T and HeLa cells (data not shown), supporting the idea that presence of variant p.Gln638*, which leads to the lack of part of the C-terminal sequence, affects protein subcellular localisation independently of the cell type. This result is consistent with predictions obtained following the Scandinavian Protocol, which combines different online tools to predict protein subcellular localisation.52 53
Discussion
Since its first description in 2013,1 more than 150 SYS individuals carrying MAGEL2-truncating mutations have been published. Most publications include one or a reduced group of patients, often with scarce clinical descriptions, hampering the definition of the syndrome’s characteristics. Families, already burdened by the solitude of having an ultrarare condition, lack clear and established clinical guidelines for their treatment and follow-up. Once available, such evidence-based recommendations will help reduce inequity in healthcare and empower both families and clinicians facing such a rare disease. To develop the recommendations, we performed an extensive revision of all the SYS subjects published so far at both the phenotypic and genetic level and elaborated a comprehensive and detailed follow-up programme. Despite the comprehensive revision, to date, no clear underlying phenotype–genotype correlation was observed, with the exception of two particular variants [p.(Gln666Serfs*36) and p.(Leu708Trpfs*7)] which are associated with the much more severe phenotype of AMC leading to perinatal death.
Genetically, SYS is caused by non-sense or frameshift mutations. Only one MAGEL2 missense variant (c.1613C>A; p.Ala538Glu) has been reliably described as potentially disease-associated.17 While at the moment of its publication, this change was classified as ‘disease causing’ based on available in silico and frequency data, currently, updated information, including a gnomAD V3.1 Amish MAF of 0.03, supports its reclassification as ‘likely benign’ according to the American College of Medical Genetics (ACMG) guidelines.54 Clinically, the patient carrying this variant presented with DD, ASD and dysmorphic traits. While her presentation shares some traits with SYS, the high frequency of this variant in the Amish population, together with the non-specific character of these traits, would suggest that it is not the main cause of the disease. Two other missense mutations have been identified in two different patients: p.Pro564Ala and p.Ala997Thr (with two and four carriers in gnomAD v3.1.1, respectively). However, their potential causality was not further discussed and there are no clinical descriptions of the patients.10 An in-frame duplication in the paternal chromosome (p.Ser724_Ala744dup, two carriers in gnomAD v.3.1) has also been identified in a patient with a clinical presentation sharing some resemblances with SYS, but the scarcity of available information makes it difficult to fully understand the clinical role of this variant.21 We have also noticed some missannotations, probably due to the fact that, originally, MAGEL2 was predicted to encode a 529-amino acid protein (instead of the current 1249 amino acids) lacking the N-terminal domain (hg38). This is the case for reported variants p.(Thr76Hisfs*23) and p.(Glu131Serfs*17),10 which we have relabelled as p.(Thr679Hisfs*23) and p.(Glu734Serfs*17). To sum up, while the pathogenicity of MAGEL2-truncating mutations has been clearly established and documented, it remains to be clarified whether any MAGEL2 missense or in-frame mutation is really a causal variant for SYS.
The phenotypic overlap between SYS and PWS suggests that the alteration of MAGEL2 may contribute to some aspects of the PWS phenotype, but the extent of this contribution is still an open question. Two patients carrying atypical deletions involving only MAGEL2, NDN and MKRN3 did not show a full PWS phenotype: one patient showed only mild delayed motor skills and the other displayed obesity, DD and high pain threshold.55 56 At the same time, neither does the deletion lead to SYS, further supporting that it is the presence of the truncated form of the MAGEL2 protein that leads to some of the particular aspects of SYS. As our results show, the truncated protein is synthesised, it is stable and it is not degraded any faster than the WT form in the first 12 hours. We propose that this truncated form could be exerting toxic effects in addition to the negative effects caused by the lack of WT MAGEL2 protein.
The role of MAGEL2 in the regulation of endosomal protein trafficking and recycling has been widely studied.41 42 57 Consistent with this, loss of paternal MAGEL2 expression in Magel2pΔ/m+ mice and neuronal cell models derived from patient with PWS leads to decreased levels of secretory granule proteins, which lead to reduced levels of circulating bioactive hormones and of mature secretory granules in neurons.36 Also, dental pulp stem cell-derived neurons from several patients with PWS and one patient with SYS showed impaired trafficking of M6PR (cation-dependent mannose-6-phosphate receptor), indicating impaired endosome-mediated retrograde transport. We hypothesised that this impaired trafficking could also be reflected in an aberrant Aβ peptide production and exocytosis.48–50 Indeed, Aβ1-40 peptide extracellular levels of SYS fibroblasts were significantly decreased compared with those of PWS and control groups, which showed no difference between them. This result supports the idea that the truncated form of the protein may have a different effect on protein trafficking than the complete loss of the protein itself and establishes Aβ1-40 levels as a potential biomarker for the disease in patient-derived cells.
Despite being its most deeply studied function, dysregulation of protein trafficking and recycling is not the only consequence of MAGEL2 malfunction. The subcellular localisation of the heterologously expressed full MAGEL2 protein41 42 and its C-terminal part alone39 has been previously studied, both presenting a mainly cytoplasmic localisation. Our immunocytochemistry assays in different transfected cell lines showed that, in contrast, the heterologously expressed MAGEL2-Gln638* (N-terminal part of the protein) was predominantly located inside the nucleus. While the lack of a properly functioning MAGEL2 antibody hampers the validations of these experiments in the endogenous MAGEL2, they point to new functions of the truncated protein. The increased nuclear localisation of the protein suggests that, in addition to the loss of the MAGEL2 normal functions, including the translocation of YTHDF2 to the nucleus,39 SYS-associated mutations could involve new functions of unknown consequences.
To explore if the truncated form of MAGEL2 could be affecting the expression of other genes, we applied an unbiased transcriptomic approach in fibroblasts. We found an enrichment in genes related to mitosis, nuclear function and localisation, as well as in developmental and neurological processes.
Previous studies on the full-length and C-terminal parts of the MAGEL2 protein have shown that this protein is involved in RNA metabolic processes.39 In our transcriptomic analysis, HOTAIR expression was clearly increased in SYS fibroblasts. HOTAIR is a well-known long non-coding RNA that mediates transcriptional silencing in trans. This gene’s promoter contains binding sites for numerous transcription factors, including AP1 (activator protein 1), Sp1 (specificity protein 1) and NF-κB (Nuclear Factor Kappa B Subunit 1).58 It is able to exert epigenetic functions through H3K27 trimethylation and H3K4 demethylation, among other regulatory functions, and has a widely studied role in cancer (reviewed in Ref. 59). HOTAIR promotes transcriptional silencing of the HOXD locus, which plays an essential role in determining body axes and orchestrating organ formation during vertebrate development.60 HOTAIR is also a regulator of mTOR, increasing its phosphorylation and mTOR-mediated exosome release,61 and its regulation of GLUT1 levels (figure 4).62 While our mRNASeq data do not show an increase in mTOR levels, SYS-derived fibroblasts have been reported to show an increase in mTOR expression and activity.63

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematisation of the relationship between the different biomarkers identified in the Schaaf-Yang syndrome (SYS) fibroblasts. The increased levels of HOTAIR could lead to the upregulation of mTOR (mammalian target of rapamycin) previously seen in the literature as well as the deregulation of glucose uptake through GLUT1 (glucose transporter 1). Ac.CoA, Acetyl-coenzyme A; GDH, glutamate dehydrogenase; SIRT4, sirtuin 4; SNAP23, Synaptosome Associated Protein 23.
Endocrine and metabolic alterations in PWS have been widely studied.64 However, little is known regarding metabolic impairment in SYS. A study including five female and four male patients with SYS showed that they may present some but not all the endocrine alterations also observed in PWS: patients benefit from growth hormone (GH) therapy, show increased ghrelin levels and present a high risk of developing diabetes mellitus.10 A review on the literature on endocrine abnormalities in MAGEL2-related syndromes26 showed that the most common hormonal alterations involve GH, thyroid-stimulating hormone, adrenocorticotropic hormone, antidiuretic hormone and gonadotropins, probably caused by hypothalamic impairment.
The targeted metabolomic analysis of SYS-derived and PWS-derived fibroblasts was aimed at finding biomarkers that assess the lysosomal, peroxisomal, mitochondrial and cytosolic metabolic pathways. The most relevant finding was a significant reduction in glutamine levels in SYS fibroblasts compared with the other two groups, whereas glutamate levels remained unchanged. Glutamine supplies nitrogen and carbon for biosynthetic reactions in rapidly proliferating cells,65 but in many contexts its key role relies on providing glutamate (figure 4) which has a wider range of metabolic functions than glutamine itself. An example of the relevant functions of glutamine and glutamate is the glutamate/GABA-glutamine cycle in neurons.66 In fact, it has been stated that alterations of the GABAergic system may play an important role in aspects of the pathophysiology of PWS.67 A hypothesis to explain the decrease in glutamine levels in SYS fibroblasts could be a dysregulation in the retrieval and recycling of a glutamine transporter, such as SLC1A5, whose retrieval and recycling is promoted by the retromer and whose degradation is enhanced on retromer knockout.68 Another explanation could be related to the hyperactivation of the mTOR pathway, as mTOR plays a role in the glutamine metabolism by increasing glutamate dehydrogenase activity through the inhibition of SIRT4 (figure 4).69 The hyperactivation of mTOR could drive a glutamate depletion which would be compensated by an increase in glutamine deamidation.
In conclusion, our results support the hypothesis that the SYS-specific phenotype might be explained by a neomorphic effect of the truncated protein, rather than by a dose-reduction situation. This is supported by the subtle changes in gene expression patterns and metabolite levels observed in fibroblasts, which suggest novel effects for the truncated protein, which warrant further exploration. In addition, we provide a comprehensive phenotypical delineation of the syndrome and a standardised set of guidelines aimed at the improvement of clinical management of patients with SYS. Finally, we propose to improve SYS diagnosis by testing the pathogenicity of doubtful missense mutations through the in vitro overexpression system.
An extended version of the methodology is included as online supplemental material. Briefly, we performed a systematic review of the literature indexed in PubMed from the date of the first clinical description of pathology associated with variants in MAGEL2 until February 2022 using the terms: ‘MAGEL2’, ‘SYS’ and ‘Schaaf-Yang syndrome’. All mutations have been referenced to the MAGEL2 hg38 main transcript NM_019066.5. Fibroblasts were obtained from skin biopsies of 7 SYS individuals, 9 patients with PWS and 11 controls (online supplemental table 6). All cell cultures (fibroblasts, HEK293T, HeLa and SAOS-2) were cultured in standard cell culture conditions. The cDNA sequences of interest were cloned in the mammalian expression vector pcDNATM3.1(+) (Invitrogen, ThermoFisher Scientific) including a HA tag and transfected into the different cell lines. Cells were treated with MG132, bafilomycin or cycloheximide. Whole RNA was extracted from fibroblasts and RNA-Seq was performed by LEXOGEN and analysed with the ExpHunter Suite.70 Expression levels of four selected genes were analysed by qPCR. Protein was extracted using RIPA buffer, resolved by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and immunoblotted following standard biochemical techniques. For immunocytochemistry, cells were fixed in 4% paraformaldehyde (PFA), permeabilised and blocked. Coverslips were incubated with anti-HA primary antibody and DAPI. For ELISA quantification, the supernatant of the collected media (without FBS (fetal bovine serum)) was obtained and analysed by the Aβ1-40 high-sensitive ELISA or the Aβ1-42 high-sensitive ELISA (IBL International). For metabolomics, amino acids were analysed using ultrahigh-performance liquid chromatography–tandem mass spectrometry and organic acids with gas chromatography–mass spectrometry. Statistical analysis was performed using R-Studio. Differences were considered significant if p<0.05.
All skin donors or their legal representative gave their written informed consent. Their samples and data were obtained in accordance with the Declaration of Helsinki 1964, as revised in October 2013 (Fortaleza, Brazil).
Abstract translation
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
The study was approved by the Institutional Review Board (IRB00003099) of the Bioethical Commission of the University of Barcelona (5 October 2020) and Hospital Sant Joan de Déu (PIC-111-19).
Acknowledgments
We would like to thank the patients’ associations Associació Síndrome Opitz C, AESYS (Asociación Española del Síndrome Schaaf-Yang) for their generous financial support and Associació PWS-Catalunya and all the families who kindly donated samples. We thank Monica Cozar for her technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
LC-V, MC-P and MS are joint first authors.
RR, SB and RU are joint senior authors.
Twitter @krabionet, @Roser_uf
Contributors MC-P, AP-P, RR, DG and RU gathered and reviewed all the clinical and mutation data previously published and elaborated the corresponding figures and tables. HF-V, LC-V, MC-P, RMC, SB and GM performed and analysed the heterologous expression experiments, WB and ICC. HF-V, RM-C, LC-V, MC-P and RU performed amilod Beta experiments. RA, CO, AJP-F, MC-P and RU performed and analyzed the metabolomics studies. LC-V, ER, JAGR, DG, SB and PS performed and analysed the mRNASeq experiments and further analysis. LCV, MS, RR, SB and RU drafted the manuscript. RU acts as a guarantor for this manuscript. All authors have critically reviewed and approved the manuscript.
Funding This research was funded through the Spanish Ministerio de Ciencia e Innovación (SAF2016-75946R and PID2019-107188RB-C21), the Instituto de Salud Carlos III-CIBERER (ACCI19P2AC720-1) and donations from Asociació Síndrome Opitz C and Asociación Española del Síndrome Schaaf-Yang (AESYS). MC-P is supported by a Carmen de Torres fellowship from IRSJD. Funding sources were not involved in the study design, collection, analysis and interpretation of data, writing of the report or in the publication of the article.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.