Article Text

Abstract
Background Axenfeld-Rieger syndrome (ARS) is characterised by typical anterior segment anomalies, with or without systemic features. The discovery of causative genes identified ARS subtypes with distinct phenotypes, but our understanding is incomplete, complicated by the rarity of the condition.
Methods Genetic and phenotypic characterisation of the largest reported ARS cohort through comprehensive genetic and clinical data analyses.
Results 128 individuals with causative variants in PITX2 or FOXC1, including 81 new cases, were investigated. Ocular anomalies showed significant overlap but with broader variability and earlier onset of glaucoma for FOXC1-related ARS. Systemic anomalies were seen in all individuals with PITX2-related ARS and the majority of those with FOXC1-related ARS. PITX2-related ARS demonstrated typical umbilical anomalies and dental microdontia/hypodontia/oligodontia, along with a novel high rate of Meckel diverticulum. FOXC1-related ARS exhibited characteristic hearing loss and congenital heart defects as well as previously unrecognised phenotypes of dental enamel hypoplasia and/or crowding, a range of skeletal and joint anomalies, hypotonia/early delay and feeding disorders with structural oesophageal anomalies in some. Brain imaging revealed highly penetrant white matter hyperintensities, colpocephaly/ventriculomegaly and frequent arachnoid cysts. The expanded phenotype of FOXC1-related ARS identified here was found to fully overlap features of De Hauwere syndrome. The results were used to generate gene-specific management plans for the two types of ARS.
Conclusion Since clinical features of ARS vary significantly based on the affected gene, it is critical that families are provided with a gene-specific diagnosis, PITX2-related ARS or FOXC1-related ARS. De Hauwere syndrome is proposed to be a FOXC1opathy.
- congenital, hereditary, and neonatal diseases and abnormalities
- eye diseases
- genetics
- genetics, medical
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Data availability statement: all data relevant to the study are included in the article or uploaded as supplementary information. PITX2 and FOXC1 variants identified in this study were submitted to ClinVar.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- congenital, hereditary, and neonatal diseases and abnormalities
- eye diseases
- genetics
- genetics, medical
WHAT IS ALREADY KNOWN ON THIS TOPIC
Axenfeld-Rieger syndrome (ARS) has been recognised for years, but due to overlapping ocular features, the term now describes two distinct genetic conditions caused by PITX2 or FOXC1 variants, with disparate non-ocular findings.
WHAT THIS STUDY ADDS
Through analysis of a large cohort of individuals with ARS, we were able to clarify the non-ocular features and identified phenotypic expansions for both conditions, especially FOXC1. We also identify De Hauwere syndrome as a type of ARS.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
We propose use of more distinct nomenclature to clearly identify for families which type of ARS their child has (PITX2-related ARS and FOXC1-related ARS) and also provide gene-specific clinical management plans. This study will have a significant impact on affected families by reducing the confusion currently surrounding ARS and guide clinicians’ medical management of these conditions.
Introduction
Axenfeld-Rieger syndrome (ARS) is a well-known ocular disorder characterised by a combination of typical ocular features including posterior embryotoxon, iridocorneal adhesions, iris hypoplasia and/or corectopia/polycoria, with or without systemic anomalies affecting the teeth, umbilicus, hearing and heart.1 2 Initially described in the 1920s–1930s and defined as separate conditions of Axenfeld or Rieger anomaly,3 4 depending on the specific combination of ocular anomalies present, the unifying term ARS was proposed in 1983 in recognition of the frequently overlapping, often syndromic and variable presentations within families.5 With the discovery of distinct genetic causes, ARS was split into subtypes.
ARS type I (MIM 180500) is characterised by ocular features, along with highly penetrant dental (hypodontia/oligodontia and microdontia) and umbilical (redundant umbilical skin/hernia and omphalocele) anomalies and is caused by heterozygous variants in PITX2. PITX2 is a transcription factor with a homeodomain that binds bicoid sequences in regulatory DNA to control expression of downstream targets.6 Pathogenic variants in PITX2 include whole gene, partial gene and/or regulatory region deletions, as well as alleles leading to the creation of premature termination codons (PTCs; nonsense, splicing and frameshift) throughout the gene and missense variants which primarily affect the homeodomain or surrounding region.
ARS type II (MIM 601499) was identified based on reports of Rieger anomaly in individuals with large 13q deletions with or without retinoblastoma and one family with linkage to 13q14.7 No single gene variants have been identified within this region, so the existence of ARS type II remains unclear and will not be further discussed here.
ARS type III (MIM 602482) has the most variable presentation, with isolated ocular defects or typical systemic features of congenital heart defects (CHDs) and hearing loss but additional anomalies occasionally reported. ARS type III is caused by heterozygous variants in FOXC1 which encodes a transcription factor with a forkhead domain that binds DNA.8 Pathogenic variants in FOXC1 include whole gene deletions or duplications, PTC variants (nonsense and frameshift) throughout the gene and missense variants within the forkhead domain and surrounding region.
The use of the term ARS to unite conditions with overlapping ocular but disparate systemic anomalies has led to substantial confusion for families. This paper reports comprehensive review of clinical features in new (81) and previously reported (47) individuals from 91 families, the largest collection of individuals with PITX2 and FOXC1 variants presented to date, to provide a better definition of the clinical spectrum for these two conditions.
Materials and methods
Human subjects
DNA was extracted from blood (EDTA tubes) or saliva (OrageneDNA; DNA Genotek, Ottawa, Ontario, Canada) samples by the Genomic Sciences and Precision Medicine Center or other labs following standard procedures.
Genetic testing was undertaken in our cohort of individuals with anterior segment dysgenesis and early-onset glaucoma phenotypes; 104 of these probands had a clinical diagnosis of ARS prior to genetic analysis. Overall rates of variant detection in ARS were calculated from the 104 probands referred to the study with a clinical diagnosis of ARS. The ARS cohort presented here includes all individuals (and affected family members) with PITX2 (59 individuals from 44 families (19 new individuals/15 new families)) or FOXC1 (69 individuals from 47 families (62 new individuals/41 new families)) variants, regardless of the initial clinical diagnosis. Clinical details were obtained through a combination of notes from referring clinicians, direct interview of participants and/or review of medical records; a standardised clinical history intake form reviewing all relevant systems (eye, craniofacial, hearing, dental, brain, endocrine, abdominal, genitourinary, skeletal, heart, gastrointestinal (GI), pulmonary, other and prenatal history) was completed by referring providers, clinical records review and/or through participant interview in the majority of cases. Counts for each feature were determined based on the number of individuals with specific clinical details available. Primary congenital glaucoma indicates glaucoma diagnosed in the first 2 years of life in the absence of visible anterior segment dysgenesis.9 MRI images (16 individuals) or reports (2 individuals) were collected and reviewed. Pairwise comparison was performed using χ2 analysis in Excel to compare features by gene or type of variant. General population frequencies were obtained from StatPearls10 for rare features (adjusted to X out of 5000 for statistical analysis): Meckel diverticulum, 1/50; imperforate anus, 1/5000; hypospadias, 1/250 men; CHD, 9/1000; hip dysplasia, 5/1000; congenital anomalies of the kidneys and urinary tract (CAKUT), 1/500; pectus excavatum, 1/400; and pectus carinatum, 3/5000.
Genetic analysis of human samples
The PITX2 and FOXC1 coding regions were examined as previously described11–13 via Sanger sequencing and/or research-based exome sequencing (Psomagen, Rockville, Maryland, USA) analysed with VarSeq software (Golden Helix, Bozeman, Montana, USA) or via clinical testing; exome sequencing was completed in 24 (41%) of PITX2 and 39 (57%) of FOXC1. Copy number analysis of the PITX2 and FOXC1 coding regions and PITX2 upstream regulatory region was performed using TaqMan assays and/or Affymetrix Genome-Wide Human SNP Array V.6.0, as previously described,2 14 15 Illumina Infinium Global Screening Array-24v2.0, or copy number analysis of exome sequencing data using VS-CNV (Golden Helix).
For PITX2, the reference transcript NM_153427.2 encoding for PITX2A was used to name all variants. In PITX2A, the homeodomain and OAR (otp/aristaless/rax) domain span aa 38–98 and aa 233–246, correspondingly.16 Other PITX2 isoforms include NM_153426.2 (PITX2B), with an additional coding exon upstream of the homeodomain, and NM_000325.6 (PITX2C), which uses an alternative start site. The C-terminal region, which includes all pathogenic alleles identified to date, is shared by all three isoforms. For FOXC1, all variants were named using NM_001453.2, the single isoform of FOXC1. Known FOXC1 domains include the forkhead domain (aa 77–168), nuclear localisation signal (aa 168-176), activation domains (aa 1-51 and 435–553), and inhibitory domain (within aa 215-365).8
Identified variants were compared with previously reported alleles as well as the frequency in the general population in gnomAD v2.1.1.17 In silico analysis of the variants was completed using SIFT, Polyphen2, Mutation Assessor, Mutation Taster and FATHMM-MKL predictions, as well as CADDphredhg19 and REVEL scores, all from dbNSFP V.4.1a, accessed through the Ensembl Variant effect predictor.18 Novel variants were assessed for pathogenicity using American College of Medical Genetics and Genomics/Association for Molecular Pathology criteria.19
Results
PITX2 genetic results and clinical features
Genetic screening identified pathogenic/likely pathogenic variants in PITX2 in 19 new individuals from 15 new families, including 6 novel alleles (online supplemental table 1). Combined with our previous reports, a total of 59 individuals from 44 families carried variants in PITX2 (figure 1A and online supplemental table 2).2 14 20 21 Causative variants in PITX2 were identified in 43/104 (41%) probands referred to the study with a clinical diagnosis of ARS and 37/41 (90%) of those with dental and umbilical anomalies also present. Individuals ranged in age from 2 months to 66 years with an average of 22 years.
Supplemental material

Schematic of PITX2 (A) and FOXC1 (B) proteins. Positions of previously reported (thin arrows) and novel (large arrows with variant specified) variants associated with Axenfeld-Rieger syndrome and related conditions indicated: circle atop arrow indicates missense/in-frame indel variants; arrows without circles indicate truncating/splicing variants; diamond atop arrow indicates both variant types present at this position. Domains indicated with shading include homeodomain (aa 38–98) and OAR domain (aa 233–246) for PITX2 and forkhead domain (aa 77–168), nuclear localisation signal domain (aa 168–176), activation domains (aa 1–51 and 435–553) and inhibitory domain (within aa 215–365) for FOXC1.
Ocular anomalies were reported in all and presented with typical ARS features in most (55/59) with additional ocular complications in some cases; the four remaining individuals were diagnosed with Peters anomaly (2) or aniridia (2) (figure 2 and table 1). Glaucoma was present in 23/32 with data available. While glaucoma onset in the first 2 years of life was occasionally seen (4), diagnosis in childhood, teens or adulthood was more common. Among individuals age 10 or older, 7/16 (44%) had been diagnosed with glaucoma by age 10; among those ≥20, 8/15 (53%) had glaucoma by 20; among those ≥49, 7/8 (88%) had glaucoma by 49. In most individuals with available treatment information, surgical intervention was required to control intraocular pressure (14/20).

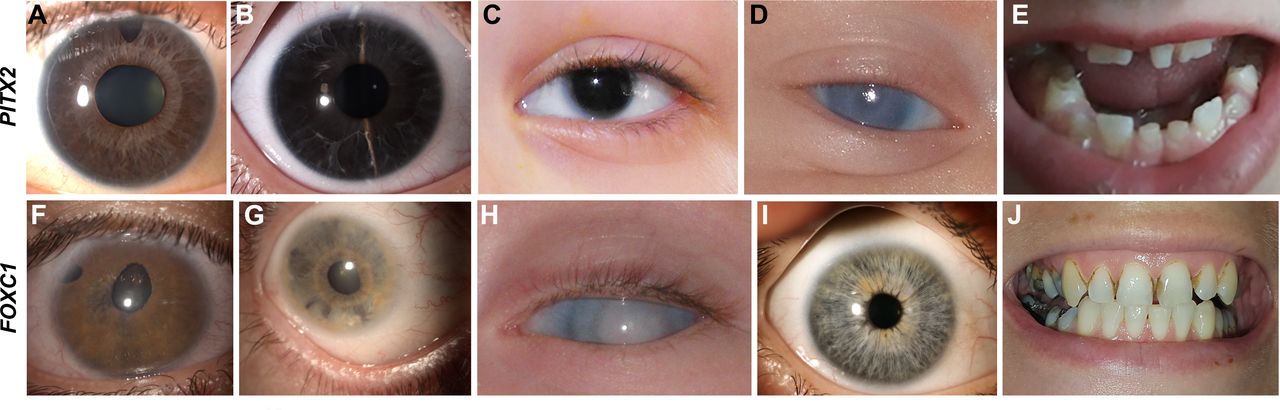
Clinical images from individuals with PITX2-related ARS and FOXC1-related ARS. (A–E) Photographs from individuals with PITX2 variants. (A) Individual 7 with ARS showing posterior embryotoxon, iris hypoplasia and polycoria. (B) Individual 14 with ARS showing iris hypoplasia and posterior embryotoxon. (C) Hendee et al 20 patient 1A showing aniridia. (D) Reis et al 2 case 21 showing corneal opacity. (E) Dental images of individual 14 showing missing teeth and microdontia. (F–J) Photographs from individuals with FOXC1 variants. (F) Individual 42 with ARS showing corectopia, polycoria and iris hypoplasia. (G) Individual 32 showing iris hypoplasia, posterior embryotoxon and iridocorneal adhesions. (H) Individual 75 with generalised anterior segment dysgenesis, corneal opacity and glaucoma diagnosed shortly after birth. (I) Individual 47 showing absence of ASD. (J) Dental image of individual 47 showing normal size and shape but poor condition with numerous caries. ARS, Axenfeld-Rieger syndrome.
Summary of clinical features observed in individuals with PITX2-related ARS or FOXC1-related ARS in this cohort
Dysmorphic facial features were reported in 37/40 and were consistent with those previously described,22 with maxillary hypoplasia and thin upper lip the most common. Additional systemic anomalies were present in all individuals with data available (57/57), with dental (40/44) and characteristic umbilical (49/52) anomalies almost fully penetrant. Dental anomalies consisted of microdontia (small and often mis-shapen teeth) and hypodontia/oligodontia (congenitally missing teeth), as previously reported; however, four individuals had generally normal dentition, perhaps somewhat small but with normal appearance and number. Meckel diverticulum (and overlapping presentations such as intestinal obstruction (1) or diverticulitis (1)) was the most common GI anomaly (15/57) with imperforate/anterior anus (3/57) and functional anomalies (irritable bowel syndrome, constipation, gastro-oesophageal reflux disease/oesophageal anomalies and gallbladder disease) also reported. While Meckel diverticulum is somewhat common in the general population, the rate was substantially increased in individuals with PITX2 variants (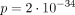 ); although rare, the rate of imperforate anus (2) was similarly increased (
); although rare, the rate of imperforate anus (2) was similarly increased ( ). Other rare birth defects increased compared with the general population included hypospadias (2,
). Other rare birth defects increased compared with the general population included hypospadias (2,  ), CAKUT (2,
), CAKUT (2,  ) and CHD (2 intragenic;
) and CHD (2 intragenic;  ); given the low frequency, it is unclear whether these increases in rare birth defects are truly associated with PITX2-related ARS or found more often due to increased screening in individuals with ARS. Consistent with the previously reported role in pituitary function, mild short stature (7) and abnormal thyroid function (3) was noted. Skeletal/joint anomalies included hypermobility/joint degeneration (4) or scoliosis (1). Developmental delay/cognitive impairment was seen in some (10); however, cognitive impairment occurred most often in individuals with larger PITX2 deletions or possibly secondary to other causes.
); given the low frequency, it is unclear whether these increases in rare birth defects are truly associated with PITX2-related ARS or found more often due to increased screening in individuals with ARS. Consistent with the previously reported role in pituitary function, mild short stature (7) and abnormal thyroid function (3) was noted. Skeletal/joint anomalies included hypermobility/joint degeneration (4) or scoliosis (1). Developmental delay/cognitive impairment was seen in some (10); however, cognitive impairment occurred most often in individuals with larger PITX2 deletions or possibly secondary to other causes.
Images from brain MRI were able to be reviewed for two adults with PITX2 disruption (table 2): one was normal, and in the other, a few punctate white matter hyperintensities (WMHs) were noted in the frontal and parietal subcortical regions (figure 3). A thin corpus callosum was also reported for one additional individual with a PITX2 deletion, but the images/report were not available for review.

Images from brain MRI from Individuals with PITX2-related ARS and FOXC1-related ARS. (A) Individual 7 with PITX2 splicing variant showing a small number of punctate T2 hyperintense lesions in frontal and parietal subcortical WM. (B–D) Individual 32 with FOXC1 deletion showing numerous ill-defined T2 hyperintense lesions mainly in subcortical WM but also periventricular and deep WM (B), mild colpocephaly (C) and bilateral staphylomas with widened perivascular spaces (C,D). (E,F) Individual 33 with FOXC1 deletion showing dysgenesis of the corpus callosum and Dandy-Walker malformation. (G–I) Individual 41 with FOXC1 nonsense variant showing numerous ill-defined T2 hyperintense lesions in periventricular, deep and subcortical WM and mild colpocephaly (G), mild volume loss of the posterior corpus callosum (H) and small anterior temporal arachnoid cyst at left as well as B staphyloma (I). (J) Individual 47 with FOXC1 frameshift variant showing a small number of punctate T2 hyperintense lesions in frontal and parietal subcortical WM and enlargement of lateral ventricles. (K) Individual 54 with FOXC1 frameshift variant showing tortuosity in the right cavernous internal carotid artery. (L,M) Individual 76 with FOXC1 missense variant showing numerous ill-defined T2 hyperintense lesions in periventricular, deep and subcortical WM and mild colpocephaly (middle childhood stage, L) and very shallow anterior chambers (neonatal birth period, M). ARS, Axenfeld-Rieger syndrome; WM, white matter.
Summary of MRI/MRA findings for individuals with PITX2-related ARS and FOXC1-related ARS
FOXC1 genetic results and clinical features
Genetic screening identified pathogenic/likely pathogenic variants in FOXC1 in 62 new individuals from 41 new families, of which 17 variants were novel (online supplemental table 1). Combined with our previous reports, a total of 69 individuals from 47 families with variants in FOXC1 were identified (figure 1B and online supplemental table 3).2 23 Causative variants in FOXC1 were identified in 31/104 (30%) probands referred to the study with a clinical diagnosis of ARS and 16/25 (64%) of those with CHD or hearing loss also present. Individuals ranged in age from 2 weeks to 62 years, with an average of 18 years.
The range of ocular features was broad. Typical ocular phenotypes of ARS, alone or in combination, were reported in 49/68, with additional ocular anomalies in some; the remaining individuals were diagnosed with aniridia (4), corneal opacity (3), aniridia with corneal opacity (3), primary congenital glaucoma (2), iris/retinal coloboma (1) or normal anterior segment (6) (figure 2 and table 1). Glaucoma was present in 38/58 individuals with data available, with more than half of individuals diagnosed within the first 1–2 years of life (25/38) and another fifth diagnosed between 2 and 10 years (8/38). Overall rates of glaucoma were similar among all age groups: among individuals age 10 or older, 14/23 (61%) had been diagnosed with glaucoma by age 10; among those ≥20 years, 11/18 (61%) had glaucoma by 20; among those ≥49, 3/6 (50%) had glaucoma by 49. In most individuals with treatment information available, surgical intervention was required (27/34).
Dysmorphic facial features were reported in 42/54 and were consistent with those previously noted,22 with widely spaced eyes (hypertelorism, broad nasal bridge and/or telecanthus) and ear anomalies reported most often. Additional systemic anomalies were present in the majority (45/65). Systemic anomalies were highly variable, both within and between families. CHD (septal defects, valve anomalies and stenosis/anomalous pathway) and hearing loss were commonly observed, seen in 21/65 each. Hearing loss occurred at all ages (figure 4)—≤2 years (4), early childhood (4), adolescence (4) or adulthood (7)—and progression over time was often noted; for those with type specified, both mixed (sensorineural and conductive) and sensorineural hearing loss were reported as well as two cases of Meniere disease. Rates also increased with age: 8/27 (30%) 10-year-olds had been diagnosed with hearing loss compared with 12/20 (60%) 30-year-olds and 8/12 (67%) 40-year-olds.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comparison of features in PITX2-related ARS and FOXC1-related ARS. (A) Genotype–phenotype correlations in ARS shows a distinct pattern of features associated with PITX2 (blue) versus FOXC1 (orange). (B) Comparison of glaucoma age of onset shows significantly earlier age of onset for FOXC1. (C) Analysis of hearing loss among individuals with FOXC1-related ARS shows onset throughout the life span but significantly higher rates for those with deletions compared with intragenic and also higher rates for PTC compared with missense alleles. ARS, Axenfeld-Rieger syndrome; n.s., not significant; PTC, premature termination codon. * p <0.05; ** p<0.01; *** p<0.001
Previously under-recognised features that were seen in a significant portion of individuals in this study include brain anomalies (reviewed further), skeletal/joint anomalies (16/65), poor growth and/or feeding disorders (16/65), CAKUT (5/65), developmental delays (19/65) and unique dental phenotypes (21/50). Skeletal anomalies included hip anomalies (7) with dysplasia/dislocation in 5, scoliosis (2), pectus deformity (2) and joint hypermobility/pain (11). Although numbers were low, the rates of hip dysplasia (p=9×10−6), pectus anomalies (p=0.00016) and CAKUT were increased compared with the general population, while rates of scoliosis were similar. Feeding disorders sometimes required use of a g-tube, and structural GI anomalies included one individual born with a tracheoesophageal fistula, which has not been reported previously, an individual with oesophageal stricture and one with a narrowed pylorus. Developmental delays resolved with time in most of the 19 cases: cognitive impairment was only noted in 6 of these individuals, generally with larger deletions (>2 Mb; 3/6) or a likely secondary cause (2/6). The classic dental and umbilical features of PITX2-related ARS were largely absent within the FOXC1 cohort, however mild redundant umbilical skin was occasionally noted (6/65) and a unique dental phenotype consisting of enamel hypoplasia/frequent caries (15/50) and/or dental crowding (8/50) was discovered.
Images/reports from brain MRI were reviewed for 16 individuals with FOXC1 disruption and showed WMH in the periventricular, subcortical and/or deep white matter in all but 1 and colpocephaly or ventriculomegaly in all but 3 (table 2 and figure 3). WMHs were stable in individuals with follow-up MRI 1–2 years from initial scan (2). Arachnoid cysts were seen in five; a variant of Dandy-Walker malformation with dysgenesis of the corpus callosum was seen in one; and two had low-lying cerebellar tonsils. Abnormal brain imaging results were noted for three out of four other individuals with completed imaging but without direct access to images/reports: gliosis/ectasia of lateral ventricles, thalamostriate vasculopathy and mild hydrocephalus. Overall, no pattern was evident between the severity of the common imaging anomalies and the presence of developmental delays/cognitive impairment. MR angiography was available for two individuals and showed vascular anomalies in both (table 2).
Genotypic spectra and genotype–phenotype correlations
For PITX2, PTC alleles were most common: 8 individuals with coding deletions, 2 with upstream regulatory region deletions, 34 with PTC variants (nonsense, splicing and frameshift) and 15 with missense variants were identified (online supplemental table 2). Average age varied between the groups: 23 years for deletion variants, 18 years for PTC and 29 years for missense. PTC variants were scattered throughout the gene, while missense variants clustered in the homeodomain (figure 1A). The type of PITX2 variant (deletion, PTC or missense) had no significant association with any of the major phenotypes (ARS ocular features, dental, umbilical or Meckel diverticulum), and all three groups showed similar rates of glaucoma. However, both cognitive impairment (p=0.025) and CHD (p=0.0025) were more common in individuals with PITX2 gene deletions bigger than 3 Mb, and individuals with larger deletions had more organ systems affected, suggesting that additional deleted genes may contribute to these phenotypes.
For FOXC1, variants were evenly distributed between the three variant types: 22 individuals with deletions, 27 with PTC variants (nonsense and frameshift) and 20 with missense variants were identified (online supplemental table 3). Average age was similar between the groups: 21 years for deletions, 17 years for PTC and 18 years for missense variants. Missense variants clustered within the forkhead domain, while PTC variants were scattered throughout the gene, similar to the distribution observed for PITX2 (figure 1B). Whole gene deletions accounted for a higher proportion of FOXC1 alleles (32% vs 17% for PITX2). Analysis of ocular phenotypes associated with different types of variants identified aniridia as more common in individuals with missense and deletion alleles compared with PTC (p=0.017 and p=0.010), while all other ocular diagnoses were similar among allele types. Hearing loss varied significantly between the groups, with the highest rate among individuals with deletions (p=0.005 vs intragenic) and the lowest rate among individuals with missense variants (p=0.016 vs del/PTC). The enamel hypoplasia/frequent caries phenotype was only reported in cases with deletions or PTC variants (p=0.004). CHD appeared somewhat more frequent with missense variants, though not statistically significant (p=0.095 vs del/PTC). Rates of other systemic anomalies were similar between the groups. For whole gene deletion alleles, the size of the deletion did not seem to have a significant impact on clinical features with the exception of cognitive impairment seen in 3/6 individuals with deletions >2 Mb, and cleft palate/bifid uvula, only seen in individuals with deletions >2 Mb.
The distribution of alleles in our cohort is similar to what was previously reported. Together with the novel alleles reported here, 106 different heterozygous intragenic PITX2 variants have now been identified in ARS and related conditions: 40 missense alleles (33 within the homeodomain), 2 in-frame variants (in the homeodomain) and 64 PTC variants (figure 1A and online supplemental tables 2 and 4). The majority of variants are private, with only one or two families reported, with the notable exception of the recurrent splicing variant c.253–11A>G reported in 10 families, as well as variants c.191C>T p.(Pro64Leu), c.356del p.(Gln119Argfs*36) and c.475_476del p.(Leu159Valfs*39) reported in 4 families each. Regulatory region deletions upstream of the PITX2 coding region have been identified in five independent families,2 14 24 25 highlighting the importance of including this region in clinical copy number variation analysis. The minimum deleted region spans conserved elements 5–7 (GRCh37/hg19 coordinates chr4:111848106–112037113). For FOXC1, 126 unique heterozygous intragenic variants are now reported in individuals with ARS and related conditions: 52 missense variants (49 in the forkhead domain), 3 in-frame variants (within or just outside the forkhead domain) and 71 premature truncating variants (figure 1B and online supplemental tables 3 and 5). Similar to PITX2, the majority of the variants are private, seen in only one or two families; one missense allele, c.482T>A p.(Met161Lys), was seen in four families.
Next, we compared the spectrum and frequency of ocular and systemic anomalies to identify common and distinct features between the two genes (table 1 and figure 4). In terms of ocular features, both genes showed similar phenotypic spectra with typical ARS ocular features in most and aniridia or corneal opacity in a number of cases. The range of ocular features was much broader for FOXC1, with 19/68 individuals having a different ocular phenotype, compared with 4/59 for PITX2 (p=0.002), and only FOXC1 was associated with a normal anterior segment in some individuals (6/68, p=0.02). The overall rates of glaucoma and surgical intervention were similar for both genes, but onset in the first 2 years of life occurred more frequently for FOXC1 (p=0.003).
Differences in syndromic anomalies clearly distinguished between the two genes. Hypodontia/oligodontia/microdontia (p=6×10−19), umbilical anomalies (p=3×10−19) and Meckel diverticulum (p=1×10−5) were highly specific for PITX2 variants. Dysmorphic facial features were slightly increased but not statistically significant for PITX2 (p=0.054) and widely spaced eyes were specific to FOXC1. While individuals with FOXC1 variants occasionally reported mild redundant periumbilical skin, this was often present only in early-life and may simply represent an outie belly button, which is common in small children.26 A distinct dental phenotype of enamel hypoplasia/frequent caries (p=7×10−5) and/or dental crowding (p=0.006), hearing loss (p=1×1o−5) and hip anomalies (p=0.01) were specific to FOXC1. Other syndromic features reported in both groups but more common for FOXC1 included CHD (p=0.0006), feeding difficulties (p=0.015), hypotonia/early delay without cognitive impairment (p=0.04) and joint/skeletal anomalies (p=0.04). Brain imaging identified a pattern of WMH and colpocephaly/ventriculomegaly with variable additional anomalies identified in individuals of all ages with FOXC1 disruption (insufficient numbers to compare to PITX2). It is interesting that brain MRI was ordered more often in this cohort compared with PITX2, possibly due to the increased frequency of hypotonia/early delays; some imaged individuals reported normal development but history of migraines.
Overall, within our cohort of 104 families referred with a diagnosis of ARS, 71% were explained by variants in PITX2 or FOXC1 after sequencing and copy number analysis, including the PITX2 regulatory region; the relative contributions of each gene were similar with 43/104 (41%) explained by PITX2 and 31/104 (30%) by FOXC1. Diagnostic rates for each gene increase when non-ocular anomalies are taken into consideration as pathogenic PITX2 variants were present in 90% of clinical ARS with dental and umbilical anomalies, while FOXC1 variants were seen in 64% of clinical ARS with CHD or hearing loss. The remaining 17 families (received with non-ARS clinical diagnoses) were explained more often by FOXC1 (94%) than PITX2 (6%), possibly due to the more variable clinical/systemic phenotype associated with FOXC1.
Discussion
Despite recognition of the syndrome for almost 100 years, ARS remains incompletely understood due to the rarity of the condition. Review of the literature identified that most papers cite 40%–60% as the proportion of ARS explained by variants in PITX2 or FOXC1.27 28 This statistic generally cites early studies of these genes, with incomplete screening in many individuals, or screening of broader ASD cohorts. Within our cohort, 71% of families with clinical ARS were explained by variants in PITX2 or FOXC1 with similar contributions from each gene. Identification of additional regulatory regions for PITX2 and FOXC1 is likely to further increase their contribution to ARS and related phenotypes.
As transcription factors, both PITX2 and FOXC1 are primarily localised to the nucleus and have DNA binding and transactivation abilities. Given the frequency of whole gene deletions, haploinsufficiency is a clear mechanism of both PITX2- and FOXC1-associated disease, but it is not the only mechanism. For both genes, the primary phenotype is generally consistent between deletions and intragenic variants, though penetrance varied for some features. For the majority of intragenic alleles, functional assessments identified loss of function but increased transcriptional activity is occasionally reported for both genes, PITX2 dominant negative and gain-of-function alleles have been seen, and FOXC1 duplication is also a known disease mechanism, suggesting that development is sensitive to exact dosage for these genes.16 29–34 While some of the phenotypic variability seen in ARS may be explained by varying levels of residual function,35 36 variability within families suggests that other modifiers also play a role in determining phenotypic outcome, particularly for FOXC1.
Analysis of this large cohort of individuals with ARS identified novel associations for both genes and clarified the distinct features for the two types of ARS. Due to the differing phenotypic presentations, it is important that families are provided with a diagnosis more specific than just ARS. While the existing ARS type I and type III nomenclature can be used to differentiate, this subtle distinction is often left off the diagnosis and is not included in many internet resources. We suggest including the gene name in the diagnosis to help families better understand which condition their child is affected with: PITX2-related ARS (currently ARS type I) or FOXC1-related ARS (currently ARS type III).
FOXC1-related ARS had a more variable ocular presentation, including several individuals with no ocular anomalies other than hyperopia or myopia, which are common in the general population, but overlapping systemic features. Review of the literature identified an additional individual with 6p25 deletion syndrome and isolated hyperopia as the only ocular finding.37 While most FOXC1 sequencing is ordered due to the presence of ocular anomalies, use of exome/genome sequencing as a diagnostic tool is likely to identify additional individuals without anterior segment dysgenesis. Although infantile/early childhood onset for glaucoma was seen more often with FOXC1 variants, both types, PITX2-related ARS and FOXC1-related ARS, showed similar rates of glaucoma overall (72% vs 66%, correspondingly), higher than the lifetime risk of glaucoma of 50% typically cited for ARS,5 16 27 but consistent with more recently reported ARS cohorts noting a 70%–75% rate of glaucoma38 with surgery required in 86%.
PITX2-related ARS was associated with nearly complete penetrance for dental (hypodontia/oligodontia/microdontia) and umbilical anomalies, as expected. Rare previously associated features27 including imperforate anus, CHD and growth/pituitary defects were also seen here; studies in mice support these associations as Pitx2 expression was observed in the developing heart, gut and pituitary gland.39 The high frequency of Meckel diverticulum is novel. While it has occasionally been noted in case reports, it has generally been considered a rare feature and is not noted in reviews of ARS.27 40 Animal models support this association as conditional deletion of Pitx2 in the mesenchyme resulted in Meckel diverticulum in 65% of mouse mutants.41 Presentation is often non-specific (unexplained anaemia and abdominal pain), suggesting that Meckel diverticulum may be under-reported.
The systemic phenotype associated with variants in FOXC1-related ARS was again much more variable. While hearing loss and CHD are well-recognised features, our study found additional systemic anomalies. Feeding difficulties, sometimes associated with structural oesophageal/stomach anomalies, are a novel association. The range of skeletal anomalies was broadened and was seen in all variant types, despite previous associations of hip dysplasia and anomalies of the torso skeleton primarily with 6p25 deletion2 42; a broad range of skeletal anomalies are similarly seen in animal models.43 44 Hypermobility and/or joint pain is a new association, supported by a prior study of differentially expressed transcription factors which identified a role for FOXC1 in the development of osteoarthritis.45 The most penetrant novel finding for FOXC1 was a unique dental phenotype consisting of enamel hypoplasia/frequent caries and/or dental crowding that has not been recognised previously and is likely under-reported. Review of the literature identified one previous case with enamel hypoplasia46 and occasional notes of dental crowding.28 37
De Hauwere syndrome (MIM 109120) is a rare phenotype with a total of three families reported.47–49 Previously, we reported a 1.3 Mb deletion of the 6p25 region involving FOXC1 and several other FOX genes in one of these individuals, providing the first genetic aetiology for this condition.2 48 In this report, analysis of the full range of phenotypic findings associated with FOXC1 alleles, including intragenic variants, demonstrated significant overlap with De Hauwere syndrome: characteristic features of Rieger anomaly, hearing loss, telecanthus/hypertelorism, short stature, joint hyperlaxity, hip dysplasia, hypotonia and hydrocephaly/dilated cerebral ventricles were all seen in individuals with FOXC1-related ARS. While hydrocephaly is rare with intragenic variants, it was reported in 39% of individuals with 6p25 deletion (including FOXC1),50 and hydrocephalus is also present in homozygous Foxc1(Mf1)−/− mice.43 Thus, we propose that De Hauwere syndrome is a FOXC1opathy. The variable expressivity associated with FOXC1 alleles, from De Hauwere syndrome to single organ involvement, highlights the need for identification of additional tissue-specific modifiers.
Some additional features observed in both populations included CAKUT and brain defects. Although rare, PITX2 showed an association with outflow abnormalities (hydronephrosis/pylectasis/hypospadias), whereas individuals with FOXC1 variants had abnormal kidney development. Rates were increased compared with the general population for both genes but more strongly for FOXC1, consistent with the recent identification of missense/indel FOXC1 variants within a CAKUT cohort51 and the presence of kidney anomalies in the mouse model52; pitx2 was also observed in the developing urogenital system in zebrafish.53 Three of the FOXC1 variants reported in the human CAKUT cohort which occurred just prior to or within the forkhead domain also caused additional syndromic features consistent with the expanded phenotype for FOXC1-related ARS (skeletal deformity, failure to thrive, blindness and high myopia), while the remainder of variants in the C-terminal region did not report extrarenal phenotypes.
While numbers were small, review of brain MRI from individuals with PITX2 variants, both here (2) and previously reported (2),54 suggests a possible increased rate of WMHs, though caution should be used since adults are known to have high rates in the general population55 and only adult images were able to be reviewed here (age in prior study unknown). For FOXC1, an association with abnormal brain MRI was strongly established at all ages, including nine children. WMH, cerebellar anomalies and hydrocephaly are recognised in 6p25 deletion syndrome37 42 56 57 but were rarely previously reported with intragenic variants. The high penetrance of WMH with intragenic variants in this cohort was surprising, but we found one previous study that noted identification of WMH in individuals with FOXC1 disruption including missense and frameshift variants.54 While an increased risk of stroke in families with FOXC1 variants was anecdotally noted by the prior group,54 further study is needed to confirm and quantify this risk as no history of stroke was reported within the FOXC1 population presented here. Work in animal models supports the role of both PITX2 and FOXC1 in the brain phenotypes.43 54 57 Although hypotonia/early delay or cognitive impairment was seen in some individuals, there was no visible pattern between the brain MRI findings and presence or absence of developmental anomalies with the exception of individual 33, with more severe structural brain anomalies and cognitive impairment. Since the volume of WMH was correlated with lower adult IQ/cognitive decline in one general population study,55 further investigation is needed specific to ARS to determine the clinical impact of these findings.
Given the disparate non-ocular findings, genetic testing for ARS in individuals with an ocular diagnosis is critical to direct clinical management in affected individuals. We suggest the following additional clinical screening subsequent to diagnosis of PITX2-related ARS or FOXC1-related ARS by molecular testing. For PITX2-related ARS, we suggest ocular exam with careful monitoring for glaucoma continuing through adulthood; treatment of umbilical anomalies as needed; referral for early dental evaluation/cosmetics to address missing/mis-shapen teeth; monitoring for GI symptoms with a low threshold to undertake diagnostic screening for Meckel diverticulum; monitoring of growth with screening for pituitary anomalies as needed; and screening for other rare associated birth defects (hypospadias, anal anomalies, heart and kidney defects). For FOXC1-related ARS, we suggest ocular exam with careful monitoring for glaucoma, especially early in life; hearing evaluation to be repeated at regular intervals throughout life; echocardiogram to rule out CHD; referral for dental care noting association of enamel hypoplasia and dental crowding; careful monitoring for feeding difficulties and/or hypotonia/delay with appropriate interventions as needed; evaluation for hip dysplasia and orthopaedic assessment as needed to address other joint anomalies; and screening for other rare associated problems (pectus, oesophageal or kidney defects). Brain MRI may be helpful in both conditions to better define the range and effect of anomalies. Information available at this time is not sufficient to provide more specific information based on the type of variant for each gene; however, some trends are emerging and may be strengthened by the identification of new cases. Since the phenotypic analysis presented here is based on available clinical data rather than consistent screening applied to every individual, subtle anomalies (such as enamel hypoplasia) or those that require specific imaging to identify (such as brain anomalies and Meckel diverticulum) are likely under-reported. Careful phenotyping with attention to all clinical features will be critical for identification of all pleiotropic effects of these genes and will allow for a precision medicine approach to improve patient care.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. Data availability statement: all data relevant to the study are included in the article or uploaded as supplementary information. PITX2 and FOXC1 variants identified in this study were submitted to ClinVar.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the institutional review boards of the Children’s Wisconsin (124172), Medical College of Wisconsin (PRO00031888) and General University Hospital in Prague (97/17), as well as sample collection protocols at the University of Michigan (1994-0495) and the University of Iowa (199804080). Written informed consent was provided by all participants/legal guardians, including permission for publication of clinical images, if applicable.
Acknowledgments
The authors are grateful to the patients and families who participated in these studies.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @joeleekw
Contributors Conceptualisation: EVS and JCM; formal analysis: LMR and MM; funding acquisition: EVS and PL; investigation: LMR, MM, LD, ST, JJ and MT; methodology and writing (original draft): LMR and EVS; resources: JC, HA, LZ, GL-S, RBL, JB, JL, AS, RKP, MV, PS, BW, GR, KB, AHS, DC, TMG, AVL, and JCM; supervision: EVS, LMR and PL; writing (review and editing): MM, JC, HA, LD, ST, LZ, GL-S, RBL, JB, JL, AS, RKP, MV, JJ, MT, BW, GR, KB, AHS, DC, TMG, AVL, PL, and JCM; guarantor: EVS.
Funding Funding for this study was provided by U.S. NIH grants (EY015518 and EY025718, EVS) as well as the Children’s Research Institute Foundation at Children’s Wisconsin (EVS) and 1UL1RR031973 from the Clinical and Translational Science Award programme. RKP was supported by AZV NV19-06-00189.
Competing interests BW and GR are employees of GeneDx, Inc.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.