Article Text

Abstract
Background Imprinting centre 2 (IC2) in the chromosomal region 11p15.5 regulates the monoallelic expression of imprinted genes by differential methylation of paternal and maternal chromosomes. Copy number variants in IC2 are associated with Beckwith-Wiedemann syndrome and Silver-Russell syndrome (SRS). Clinical outcome of IC2 deletions seems to depend on the parental origin of the chromosome, deletion size and inclusion or exclusion of enhancer and promoter regions.
Results A paternally inherited 132 bp deletion within the KCNQ1OT1 gene was found in a proband with an SRS clinical phenotype. The patient’s father and paternal grandmother, who both carry the deletion on their maternal chromosome, are unaffected. Review of other IC2 deletions and their associated clinical presentation was useful in understanding the genetic–phenotypic correlation.
Conclusion Only six cases have been reported with deletions involving exclusively IC2, one being identical to our proband’s 132 bp deletion. Our study, which is based on more extensive segregation data than the previous 132 bp deletion report, confirms the association of this deletion with growth restriction when paternally inherited. Remarkably, even though our patient has the same deletion, he has more pronounced phenotypic features; our findings thus suggest that some degree of clinical variability may be associated with this loss.
- Genetics
- Sequence Deletion
- DNA Methylation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Silver-Russell syndrome (SRS) is a rare inherited disorder characterised by phenotypic features such as growth retardation, facial dysmorphism and limb asymmetry.1 In this genetically heterogeneous condition, a genetic diagnosis can be made in approximately 60% of patients.1 The Netchine-Harbison Clinical Scoring System (NH-CSS) can be used to determine clinical suspicion of SRS in the presence of at least four of the following six criteria: prenatal growth retardation, postnatal growth retardation, relative macrocephaly at birth, protruding forehead, body asymmetry and feeding difficulties.2 These criteria are defined by substantial deviation from mean reference values of length, weight (≥2 SD prenatally, at birth or at 24 months) or head circumference (>1.5 SD above birth weight and/or length) at various developmental milestones. Protruding forehead is defined as one that projects beyond the facial plane when viewing from the side, and body asymmetry is a discrepancy of ≥0.5 cm in length between limbs or two or more other asymmetric body parts with limb discrepancies less than 0.5 cm. The last criterium is feeding difficulties or low body mass index at 24 months or current use of feeding tube or weight gain–promoting medication. The diagnosis requires specific molecular findings for confirmation.1
Genomic alterations or methylation defects in the 11p15.5 region can lead to either Beckwith-Wiedemann syndrome (BWS) or Silver-Russell syndrome (SRS), which have opposite growth effects.1 The genes contained in the 11p15.5 chromosomal region are regulated by two functional domains. In one of these domains, the expressions of IGF2 and H19 genes are regulated by imprinting centre 1 (H19/IGF2:IG-DMR, IC1). The other imprinting domain contains the imprinted genes KCNQ1, KCNQ1OT1, CDKN1C, SLC22A18 and PHLDA2, which are regulated by imprinting centre 2 (KCNQ1OT1:TSS-DMR, IC2). This imprinting centre overlaps exon 1 of the KCNQ1OT1 gene (NR_002728.3), part of the KCNQ1OT1 promoter region and intron 11 of the KCNQ1 gene (NM_000218.3).3 Imprinting centres regulate imprinted gene expression using selective methylation of maternally and paternally inherited chromosomes.3 IC2 regulates the maternally expressed cell–cycle inhibiting factor CDKN1C via the non-coding transcript of KCNQ1OT1.4 BWS and SRS phenotypes have been linked to genetic variants in IC2, such as deletions.4 The severity of these phenotypes depends on the size of the deletion and if the allele is maternally or paternally inherited.4
Here, we present a patient with a paternally inherited 132 bp deletion in IC2 that affects the KCNQ1OT1 gene. This exact deletion has been previously reported in another patient with growth retardation and some SRS phenotypic features such as mild prominent forehead, low set and prominent ears as well as downturned corners of the mouth, but this deletion was not associated with intrauterine growth restriction (IUGR).4 Our patient seems to have a more pronounced SRS phenotype. The deletion did not impact DNA methylation at IC2, but likely impacts the expression of imprinted genes.
Case presentation
The patient, a young boy, is the only child of healthy non-consanguineous parents. Pregnancy was achieved by spontaneous conception, and the mother underwent prenatal serum screening in the context of advanced maternal age. The latter screen showed increased risk for trisomy 21. Therefore, amniocentesis was completed at 19 weeks’ gestation, but karyotyping did not detect any abnormalities. Pregnancy was also complicated by second trimester IUGR. The patient’s mother (age 30–40) developed benign intracranial hypertension at 26 weeks of gestation. Three weeks later, the patient was born via caesarian section following poor umbilical artery flow and severe IUGR. APGAR score was 7 at 1 min and 7 at 5 min. The patient’s birth weight was 756 g (less than third percentile). Head circumference was within the normal range at 25.5 cm (25th percentile). The neonatal period was complicated by lung prematurity, hyperbilirubinaemia, feeding difficulties, germinal matrix haemorrhage, retinopathy of prematurity, heart murmur (patent ductus arteriosus) and anaemia requiring transfusions. The patient also had early hypoglycaemia, hyponatraemia and hypoalbuminaemia requiring intravenous perfusions. Mild glandular hypospadias, poor axial muscle tone and absence of head elevation were noted on physical examination. After 4 months of hospitalisation in the neonatal intensive care unit, the patient had persistent feeding difficulties and failure to thrive with head sparing.
In infancy and childhood, the patient displayed motor and speech delay, attention deficit and hyperactivity disorder, oppositional defiant disorder and learning difficulties. In addition, the family reported excessive sweating and variable appetite with no objectified hypoglycaemic episodes. The patient also had recurrent epistaxis requiring frequent cauterisations which was later attributed to a deficiency in coagulation factor VIII. On physical examination, proband height was 125.2 cm (3rd percentile), weight was 21.2 kg (3 SD below mean), and head circumference was 51.3 cm (25th percentile). The patient’s bone age was measured at 1.3 SD below the reference range. Facial features included a triangular face with frontal bossing, micrognathia, high nasal bridge and long eye lashes. The patient’s Netchine-Harbison clinical scoring for RSS is estimated at 5 out of 6, which raised suspicion for this condition and triggered molecular testing.2
Relatives on the paternal grandfather’s side have been described as short, but family history is otherwise unremarkable. The father’s height was measured at 172.5 cm (25th percentile) and the mother at 159.4 cm (10th percentile).
Materials and methods
Methylation-sensitive and dosage-sensitive PCR analysis using MS-MLPA kits ME030-C3 and ME032 (MRC Holland) were used to examine the presence and copy number of differentially methylated sites associated with SRS. ME030 detects both dosage and methylation of the imprinting control region of H19 and IGF2 at 11p15.5. ME032 detects both dosage and methylation status of two regions on chromosome 7 (GRB10 at 7p12.1 and MEST at 7p32.2) associated with SRS. The genomic sequence of the region affected by the deletion was then amplified by standard PCR using a high-fidelity PCR master mix (Sigma-Aldrich, catalogue #12-140-314-001) with 50 ng genomic DNA and analysed by Sanger sequencing. DMSO was added to the PCR (5% of final reaction) as the target region is GC-rich. The forward and reverse KCNQ1OT1 primers (5′−3′; DNA Technologies IDT), with a final concentration of 0.3 μM in the reaction, were tgtaaaacgacggccagtGGGTGGCATCAAAACSAGAC and caggaaacagctatgaccCCCGGGGAGAACAGAACC, respectively. Capital letters indicate gene-specific sequence, while small letters show M13 tails used for sequencing.
Results
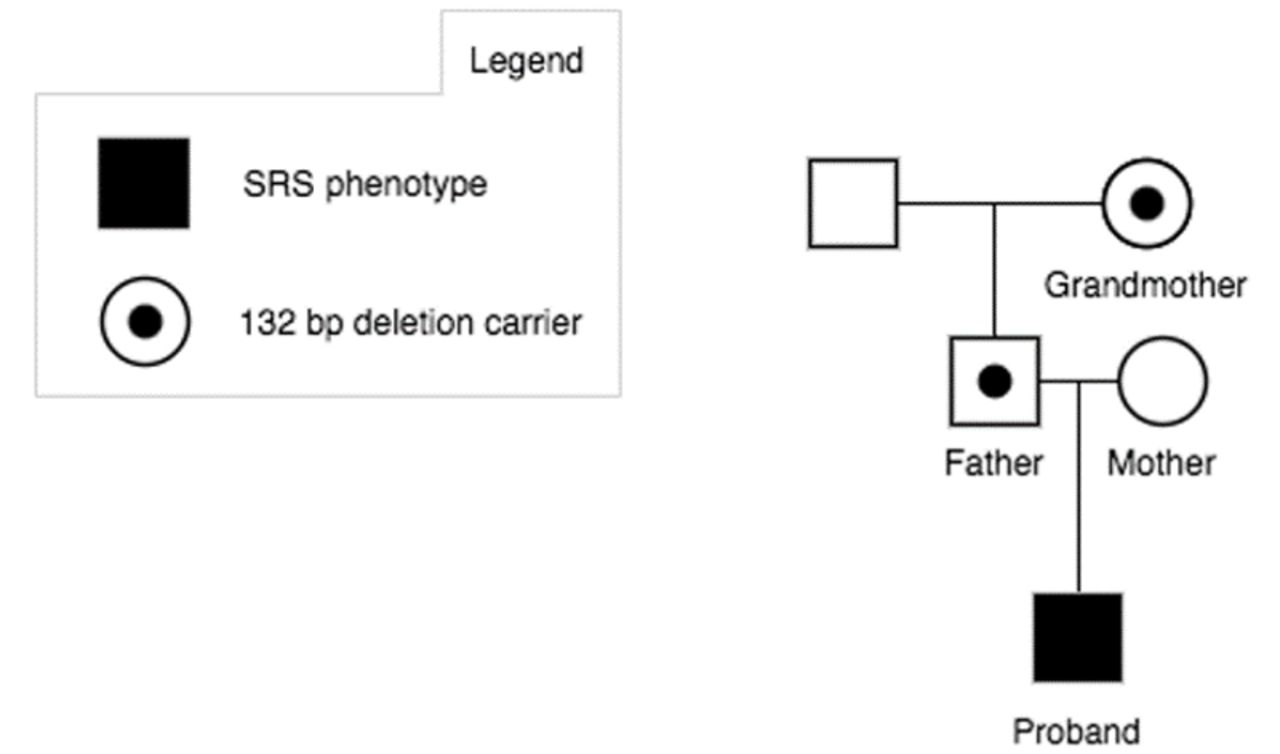
Methylation-specific multiplex ligation-dependant probe amplification (MS-MLPA) analysis was performed in the patient for SRS-associated loci at 11p15.5, 7p12.1 and 7p32.2. A normal MS-MLPA result was obtained for the loci on chromosome 7, but a heterozygous deletion was detected in the chromosomal region 11p15.5. To be more precise, it concerns a heterozygous deletion of one MLPA probe (KCNQ1OT1, 07172-L06781; ME030-C3) with a hypermethylation signature for the respective probe, which suggests the deletion resides in the paternally inherited allele (figure 1 and online supplemental figures S1-S3). Sanger sequencing of this region identified a heterozygous 132 bp deletion in KCNQ1OT1 at chr11:2720964-2721095 (GRCh37) (HGVS recommended nomenclature: NR_002728.3:n.134_265del; NC_000011.9:g.2720965_2721096del) (figure 1 and online supplemental figures S3). Parental testing identified the same deletion in the father, confirming the patient’s deletion is paternally inherited. The father, who is of normal phenotype, has hypomethylation in the same region, which corresponds to a heterozygous deletion on his maternal allele. The patient’s paternal grandmother, who is of normal phenotype, is a heterozygous deletion carrier and also has a hypomethylation signature for the respective probe, which suggests that she carries the deletion on her maternal allele. These results support an association of the 132 bp KCNQ1OT1 deletion, if paternally inherited, with growth retardation (figure 1).
Supplemental material

{kind=link}
Pedigree of the proband with a Silver-Russell syndrome (SRS) phenotype and a 132 bp deletion involving KCNQ1OT1.
Discussion
In this study, we have identified an individual with an SRS phenotype and a 132 bp deletion overlapping the KCNQ1OT1 promoter region and IC2. Less than 1% of SRS cases have copy number variations in the 11p15.5 IC2, and most of them are duplications.4 Deletions exclusively targeting IC2 in the maternal copy of genes KCNQ1OT1 and KCNQ1 have been associated with BWS, while paternally inherited deletions affecting these genes have been associated with SRS or growth restriction (online supplemental table S1).4–10 These deletions can also be observed in individuals with a normal phenotype, since paternal or maternal inheritance plays a role in disease manifestation.4
Paternally inherited microdeletions in IC2 likely lead to an increased expression of CDKN1C, a cell cycle inhibiting factor, due to the disruption of KCNQ1OT1, which is a repressor of CDKN1C.7 We therefore believe that our patient’s phenotype is caused by such a disruption. Only the paternal allele of KCNQ1OT1 is expressed and linked to growth; therefore, our patient’s father and paternal grandmother have a normal phenotype as they carry the deletion on their maternally inherited chromosomes.
It has been suggested that variability in SRS phenotype can be explained by whether regulatory regions (such as enhancers and promoters) are included or not, in deletion events near IC2. Several enhancer regions in close proximity to the KCNQ1OT1 gene are thought to regulate CDKN1C expression. It has been suggested that small deletions leading to a loss of KCNQ1OT1 leave these enhancers intact, leading to CDKN1C expression and subsequent growth suppression.10 Larger deletions that include these enhancers likely lead to a milder phenotype through disruption of positive CDKN1C regulatory elements in addition to KCNQ1OT1.8 11 This can explain why a relatively small 60 kb deletion, which likely does not include a CDKN1C enhancer regions, had a more severe phenotype with intrauterine growth failure compared with larger deletions involving one or more proposed enhancer regions.11 Our patient’s 132 bp deletion is likely too small to encompass such regions, thus causing growth suppression.
As previously reported, an identical 132 bp deletion was detected in a 5-year old girl with an SRS phenotype inherited from her father who was unaffected as he carried the variant on his maternal chromosome.4 She had postnatal growth retardation, mild prominent forehead, low set prominent ears and downturned corners of the mouth. Interestingly, this patient also had delayed bone age (16 months delay) with normal IGFBP3, IGF1 and growth hormone stimulation test. Our patient seems to have a more severe phenotype with IUGR, hypoglycaemic episodes, feeding difficulties, excessive sweating, behavioural issues and more prominent facial dysmorphism. Although our report supports an SRS phenotype linked to KCNQ1OT1 deletions, other causes of IUGR cannot be ruled out in our patient. Notably, advanced maternal age and onset of benign intracranial hypertension in the mother during second trimester may also have contributed to the growth retardation.
In summary, paternally inherited deletions affecting IC2 exclusively are rare and have variable clinical presentations. Regardless, testing for IC2 deletions in patients with SRS features is important and should be included in the diagnostic workup. Further studies on IC2 deletions could lead to a better understanding of IC2’s role in growth regulation.
Ethics statements
Patient consent for publication
Ethics approval
The present study was reviewed by the Vitalité Health Network ethics board and was approved with reference number 101330. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank the family for study participation and B. Grondin and M. Huyck for excellent technical assistance, respectively.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
MVG and EPA contributed equally.
Contributors HHA and LMG performed the MS-MLPA experiments and analysed the data. MVG and MBA reviewed the case. MVG, EPA and MBA wrote the manuscript. EPA, MBA, HHA and LMG reviewed and edited the manuscript.
Funding This project was funded by the Centre de formation médicale du Nouveau-Brunswick and the New Brunswick health research foundation.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.