Article Text

Abstract
Background Whole-exome sequencing-based diagnosis of rare diseases typically yields 40%–50% of success rate. Precise diagnosis of the patients with neuromuscular disorders (NMDs) has been hampered by locus heterogeneity or phenotypic heterogeneity. We evaluated the utility of transcriptome sequencing as an independent approach in diagnosing NMDs.
Methods The RNA sequencing (RNA-Seq) of muscle tissues from 117 Korean patients with suspected Mendelian NMD was performed to evaluate the ability to detect pathogenic variants. Aberrant splicing and CNVs were inspected to identify additional causal genetic factors for NMD. Aberrant splicing events in Dystrophin (DMD) were investigated by using antisense oligonucleotides (ASOs). A non-negative matrix factorisation analysis of the transcriptome data followed by cell type deconvolution was performed to cluster samples by expression-based signatures and identify cluster-specific gene ontologies.
Results Our pipeline called 38.1% of pathogenic variants exclusively from the muscle transcriptomes, demonstrating a higher diagnostic rate than that achieved via exome analysis (34.9%). The discovery of variants causing aberrant splicing allowed the application of ASOs to the patient-derived cells, providing a therapeutic approach tailored to individual patients. RNA-Seq data further enabled sample clustering by distinct gene expression profiles that corresponded to clinical parameters, conferring additional advantages over exome sequencing.
Conclusion The RNA-Seq-based diagnosis of NMDs achieves an increased diagnostic rate and provided pathogenic status information, which is not easily accessible through exome analysis.
- neuromuscular diseases
- diagnosis
- RNA-Seq
Data availability statement
Data are available on reasonable request. Anonymised data not provided in the article may be shared at the request of any qualified investigator for purposes of replicating procedures and results.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known on this topic
Diagnostic process of neuromuscular disorders (NMDs) poses opportunity to use muscle biopsy samples for improvement.
What this study adds
Usage of muscle RNA sequencing in NMDs enhanced diagnostic yield.
How this study might affect research, practice or policy
Transcriptome analysis on biopsy-derived muscle tissue can be used as an effective complement to conventional whole exome sequencing-based diagnostic strategy.
Introduction
Neuromuscular disorders (NMDs) affect the normal function of muscle either by directly impairing muscle structure and metabolism or by indirectly affecting signal transfer from neurons to muscles.1
NMD present with variable clinical symptoms and prognosis in which majority of disorders result in lifelong functional impairment and burden. Accurate clinical and genetic diagnosis can lead to adequate monitoring and treatment of the patient symptoms. Conventionally, NMDs are diagnosed by muscle biopsy and pathological evaluation.2 Although whole-exome sequencing (WES) has become a routine diagnostic tool in clinical practice, its diagnostic yield for NMD has remained unchanged in recent years.3–7 Even in diseases with an established genetic cause, not all cases show variants within the known pathogenic genes due to locus heterogeneity or phenotypic heterogeneity. Additionally, even carriers of a known disease allele do not necessarily display the associated phenotype (ie, incomplete penetrance).8 Therefore, transcriptome sequencing has been tested as an approach for augmenting the discovery and interpretation of additional variants9–13; however, clinical and genetic variability in samples complicates the interpretation of gene expression values.14 Moreover, the correlations between muscle pathology, clinical symptoms, prognosis and molecular signatures have yet to be clearly delineated.
To maximise diagnostic yields for NMDs, we devised a transcriptome-based pipeline devoid of exome analysis; the results demonstrate that transcriptome sequencing provides an effective independent platform for NMD diagnosis. We further illustrate the utility of RNA sequencing (RNA-Seq) data in NMD by correcting aberrant splicing events by treating patient-derived cells with antisense oligonucleotides (ASOs) and constructing comprehensive transcriptomic profiles to identify subgroups with distinct gene expression profiles.
Methods
A total of 117 patients with suspected Mendelian NMD but without a definite molecular cause were enrolled from Seoul National University (SNU) Hospital between 2000 and 2020. All cases were ascertained from the NMD database in the SNU Hospital.
All patients or their guardians provided informed consent. The histopathology of muscle-biopsied tissues was reviewed at the SNU Children’s Hospital. Muscle samples were taken from the quadriceps femoris. Muscle tissues from four healthy individuals were collected during plastic surgery procedures as control samples. The detailed clinical information of the cohort is provided in online supplemental table 1.
Supplemental material
Data collection was planned and performed after the sample collection. Whole-blood and muscle biopsy samples were used for DNA and RNA extraction as described previously.9 15 The Illumina paired-end sequencing of libraries constructed with the Agilent SureSelect Human All Exome v5 and TruSeq Stranded Total RNA Sample Prep kits was performed at Theragen Etex (Suwon, Korea). For each WES and RNA-Seq run, 40–60 million paired-end reads of 101 or 151 bp were generated. Comprehensive descriptions of procedures used for RNA-Seq data processing, variant calling and evaluation, pathogenic variant identification, aberrant splicing event discovery, ASO design and functional validation, non-negative matrix factorisation (NMF) clustering and evaluation, and tissue deconvolution are provided in the online supplemental information. Custom Python and R scripts were used to parse and visualise the results. A p-value of 0.05 was defined as significant and multiple test correction was applied using the Benjamini-Hochberg procedure.
Supplemental material
Results
Discovery of causal variants through variant calling from muscle transcriptomes
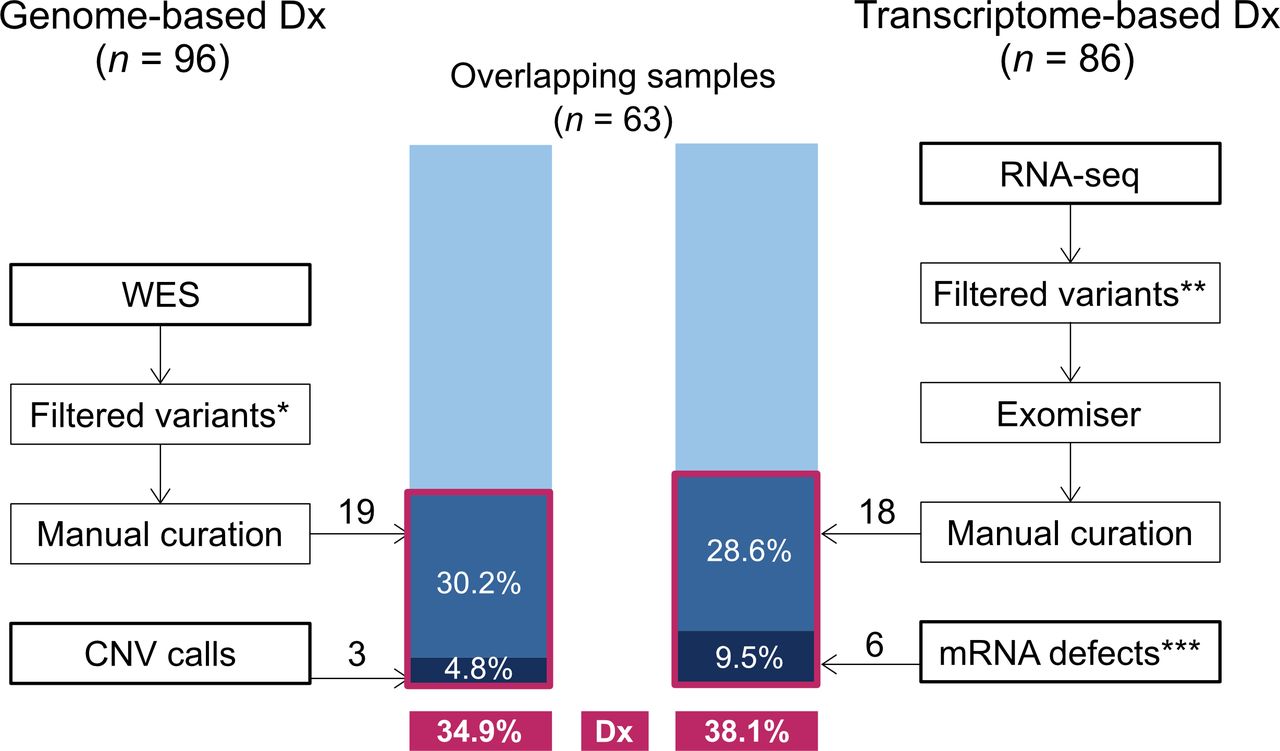
Among the tissue samples from 117 patients with NMD (online supplemental table 1), blood was subjected to WES (95 samples passed quality control (QC), 1 sampled failed QC) and muscle to RNA-Seq (86 samples). Among the 86 samples, 83 muscle transcriptomes passed the quality assessment tests and clustered with Genotype-Tissue Expression project (GTEx)16 muscle samples in a principle component analysis (PCA) plot (online supplemental figure 1) and 63 patients were analysed by both methods (figure 1).

Diagnostic (Dx) yield of neuromuscular disorders (NMDs) using transcriptome data. Diagnostic yields were compared for 63 patients with NMD who were simultaneously analysed by whole-exome sequencing (WES) and RNA sequencing (RNA-Seq) approaches. Samples in red boxes denote diagnosed samples by each approach. *Protein-altering variants with an allele frequency <0.01 and Genotype Quality (GQ) >60. **Coverage depth >5 and indel length <70. ***These include splicing variants, outlier expression and variants causing allele-specific expression.
WES yielded candidate genes in 27.4% of individuals (26/95). Of those sequenced, 50.0% (13/26) carried variants described as ‘pathogenic’ or ‘likely pathogenic’ in ClinVar17, and 46.2% (12/26) carried variants in known muscle disease genes; patients with these variants displayed symptoms matching previous reports. Of the seven variant-carrying individuals that were available for trio-WES, all had the pattern of variant segregation confirmed (six de novo and one homozygous variants). Variants in RYR1 were most common (n=6), followed by variants in COL6A2 (n=4) and ADSS1 (n=2). Deletions encompassing DMD or MICU1 were found in separate patients (online supplemental table 2).
RNA-Seq yielded candidate genes in 28.9% patient samples (24/83), including 18 from variant calling and 6 from aberrant splicing analysis. Among the six aberrant splicing events, four were found in DMD, and one case each was found in MICU1 and LPIN1. We did not identify any pathogenic allele-specific expression event. For the 63 patients who were analysed by both methods, RNA-Seq yielded an overall diagnosis rate of 38.1% while WES achieved 34.9% (figure 1).
Next, we tested the diagnostic ability of muscle transcriptome-based variant calling and compared it with exome-based variant analysis. For this analysis, we used 19 samples having both WES and RNA-Seq data, and WES-identified causal genes. In establishing a computational pipeline to call and prioritise potential pathogenic variants, we modified the standard RNA-Seq variant-calling pipeline and incorporated variant prioritisation software (online supplemental materials). We chose to test Exomiser, Divine and DeepPVP and found that the performance of Exomiser outranked those of others, as it reproduced 94.7% of WES candidate genes identified in individuals with both types of data available (18/19 calls made by Exomiser ranked among the top 10 candidates, compared with 16/19 for DeepPVP and 12/19 for Divine; the mean rank number for Exomiser calls excluding the homozygous MYO9A call was 4.1, whereas the means for DeepPVP and Divine were 21.9 and 28.3, respectively; figure 1, online supplemental table 3).18 19 The undetected homozygous variant in MYO9A (NM_006901.4:c.3791A>C) was not in the top 10 candidates according to all of the tested software due to showing a relatively high allele frequency in East Asians (0.82%, but no homozygotes were found in gnomAD). Therefore, transcriptome-based analysis yielded a higher diagnostic rate than trio-based WES analysis (figure 1).
Additional detection of aberrant transcripts from muscle
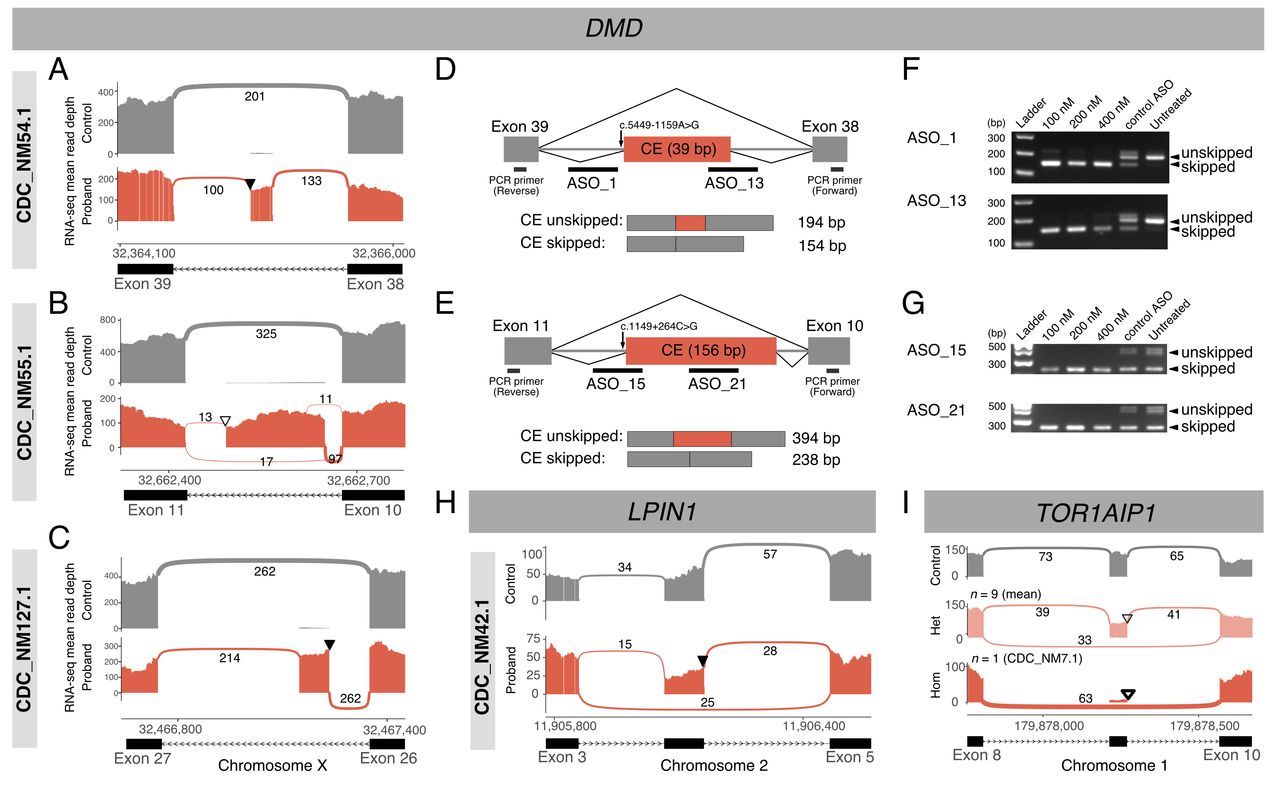
We then sought to identify additional genes that may cause or contribute to NMD from transcriptome data and noted several aberrant splicing variants. To detect outliers, we included transcriptomes obtained from patients (n=83) and normal controls (n=4). Fewer than 10 significant outlier splicing events (padj <0.05) were detected in most samples (73/87), and the manual inspection of each event and patient phenotype identified several pathogenic splicing variants (online supplemental table 4). For example, in three samples from patients showing a typical DMD-related phenotype with dystrophin loss in their pathology, no pathogenic variants were identified by WES. RNA-Seq data suggested the existence of cryptic exons (CEs) in DMD, and subsequent WGS identified maternally inherited intronic variants that may have produced aberrant splicing and resulted in premature termination (figure 2A–G). Additionally, we detected a heterozygous exon-skipping event in LPIN1 from a patient diagnosed with recurrent rhabdomyolysis; this event was due to a variant in the last base of the skipped exon, which was inherited from his mother (figure 2H). This variant was not prioritised as pathogenic by WES because it was annotated as a synonymous variant (SNV) and was not predicted to be splice-altering by SpliceAI.20 Combined with a nonsense variant inherited from the patient’s father, compound heterozygosity was observed for LPIN1, explaining his symptoms (OMIM: 268200). We also describe a notable recurrent exon-skipping event in TOR1AIP1. RNA-Seq data revealed consistent exon skipping events in one homozygote and nine heterozygotes for a splice donor site variant within our cohort (figure 2I). While the homozygous patient exhibited a TOR1AIP1-associated phenotype, we concluded that this variant was not directly causal because the variant shows a high allele frequency in East Asians in gnomAD (6.8%). Notably, among six SNVs causing aberrant splicing events, one SNV in LPIN1 and one deep intronic variant in DMD were not predicted to be splice-altering by SpliceAI software (online supplemental table 4).20

Discovery and rescue of abnormal transcripts due to splicing-altering variants. (A–C) Three cases with cryptic exons (CEs) in DMD, ultimately causing premature terminations ((A) CDC_NM54.1, (B) CDC_NM55.1 and (C) CDC_NM127.1). Whole-genome sequencing (WGS) further identified likely causative deep intronic variants. Thickness and numbers over the arcs linking adjacent exons represent the junctional reads supporting splicing events. Triangles indicate variants that colocalise with CEs ((A) NM_004006.2:c.5449–1159A>G, (B) c.1149+264C>G and (C) c.3603+820G>T). Filled triangles denote variants confirmed to be maternally inherited. Parental samples of CDC_NM55.1 were not available, and the variant is shown as an open triangle. (D and E) Splicing and antisense oligonucleotide (ASO)-targeting schemes in two patients with DMD ((D) CDC_NM54.1 and (E) CDC_NM55.1). Orange boxes denote CEs and grey boxes indicate flanking exons. Thick black horizontal lines indicate individual ASO target sites or primers used for RT-PCR, encompassing CEs. (F–G) RT-PCR products visualised by agarose gel electrophoresis ((F) CDC_NM54.1 and (G) CDC_NM55.1). (H) A case of heterozygous exon skipping in LPIN1. A maternally inherited synonymous variant is located in the last base of the exon (NM_145693.4:c.288G>A), which is denoted with a filled triangle. (I) Sashimi plots of nine heterozygotes (middle; open triangle) and one homozygote (lower; thick open triangle) of a splice donor site variant in TOR1AIP1 (NM_001267578.1:c.967+1G>A).
Therapeutic modulation of splicing defects by patient-specific ASO treatment
Next, to test whether these deep intronic variants that disrupted proper DMD splicing were therapeutically targetable, we attempted to rescue the splicing defect using ASO. Myoblast lines isolated from muscle biopsies were available from two of the patients (CDC_NM54.1 and CDC_NM55.1; figure 2D,E). ASO treatment efficiently induced CE skipping and restored the normally spliced DMD transcript, as confirmed by RT-PCR (figure 2F,G). These effects were observed at all tested ASO concentrations (100–400 nM), and ASOs targeting different positions of the CEs showed various degrees of CE removal efficiency (online supplemental figure 2).
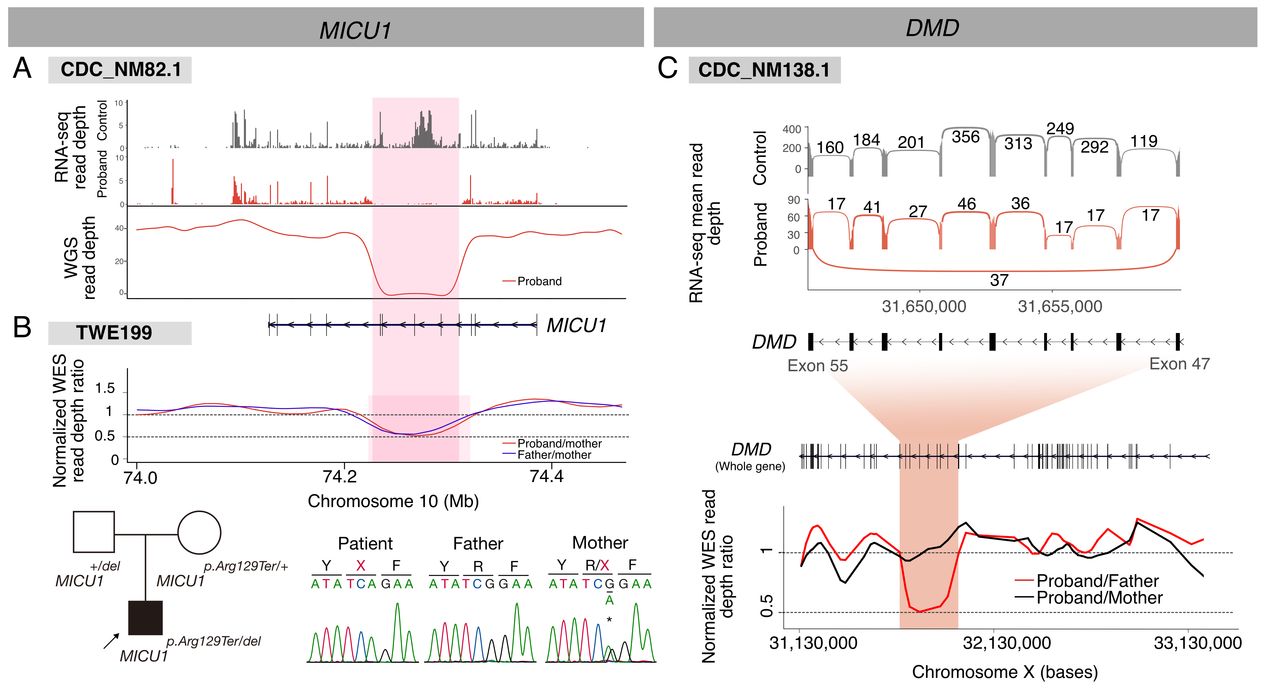
We also discovered deletion events that encompassed several exons and were not detected by singleton WES due to uneven coverage of sequencing reads. A homozygous exon-skipping event in MICU1 was associated with myopathy with extrapyramidal signs21; subsequent WGS revealed an ~82 kb homozygous deletion encompassing four exons (figure 3A; OMIM: 615673). Notably, an unrelated patient with similar symptoms was found to carry an overlapping paternal deletion, along with a maternal premature termination variant (figure 3B). These cases implied that the deletion of this region is a rare causative factor in the Korean population. Finally, a heterozygous deletion encompassing seven exons (~220 kb) in DMD was identified in a female patient in her 50s with adult-onset progressive muscle weakness (figure 3C). We did not find notable events in allele-specific expression or gene expression outlier analysis.
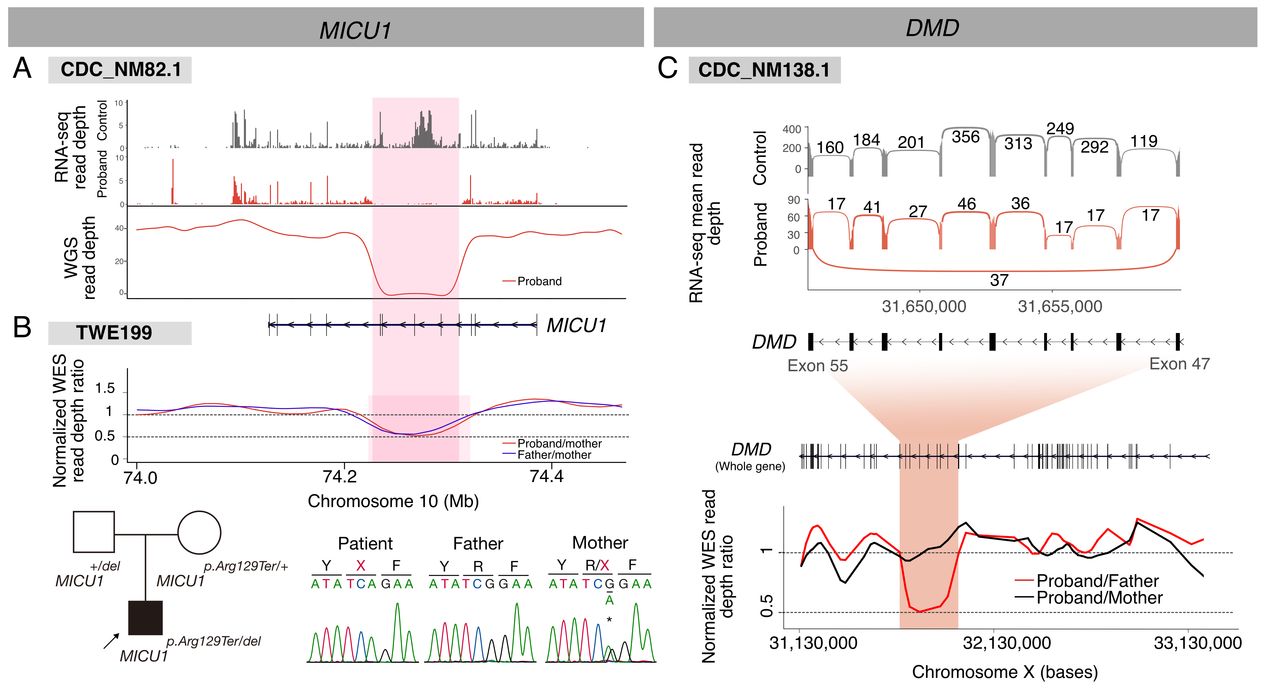
Deletion events detected by RNA-Seq. (A) A homozygous deletion in MICU1 causing a loss-of-function defect in CDC_NM82.1. (B) A similar heterozygous deletion was found in an independent patient with similar symptoms (TWE199) by trio-WES. (C) A heterozygous exon skipping event in DMD found in a manifesting female patient shown as a Sashimi plot (upper). Note a low arc connecting two extreme exons in the proband plot. A subsequent trio-WES read depth analysis (lower) identified a corresponding maternally inherited genomic deletion. RNA-Seq, RNA sequencing; WES, whole-exome sequencing.
Assessing molecular features of muscle transcriptome by expression-based clustering
In addition to diagnostic utility, RNA-Seq may provide additional benefits in assessing patients with NMD by characterising patient-specific molecular pathways and predicting therapeutic effects through expression-based clustering. We characterised transcriptomic variability of the muscle samples, which may present differential proportions among cell types. Since our RNA-Seq data were produced from muscle tissues in bulk, we attempted to deconvolute the data to obtain the contributions of each cell type. Indeed, deconvoluted cell type abundances captured the clinical and pathological differences between samples (figure 4A, online supplemental table 5). The abundances of fibroadipogenic progenitor (FAP) cells successfully predicted fibrosis (R2=0.23; p=4.9×10−6) and adipose scores (R2=0.18; p=7.5×10−5), the contribution of type IIa fibres explained the predominant fibre type (p=7.3×10−3; Wilcoxon rank sum exact test) and the proportion of satellite cells predicted biopsy ages (R2=0.57; p=1.2×10−16) (figure 4A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clustering analysis of 83 affected and four control muscle samples. (A) Scatter plots demonstrating correlations of pathological and clinical information and relative abundances of muscle cell types according to the deconvolution results in each sample. (B) Pathological and clinical features of each non-negative matrix factorisation (NMF) cluster. (C) A heatmap showing 11 distinct sample clusters generated by NMF and their relative cell type abundances. Samples with asterisks are control samples. Notable features mentioned in the text are marked in black boxes. (D) Gene Ontology (GO) profiles of selected clusters. (E) Scatter plot displaying the correlation between onset age and biopsy age across all clusters. FAP, fibroadipogenic progenitor; NK, natural killer.
Additionally, as our cohort included various clinical and genetic diagnostic categories, we hypothesised the presence of distinct sample clusters that may be represented by genes with specific functions. Transcriptome-based clustering using NMF identified 11 clusters, each of which contained 7.9 samples on average (figure 4B,C). Notably, patients with samples in cluster 8 exhibited high degrees of fibrosis and adipose replacement and were clinically classified as ‘slowly progressive but with prolonged muscle disease’; in this cluster, FAP cells were relatively abundant (p=2.7×10−5; Welch’s t-test) and lipid metabolism-related gene expression was significantly altered (padj=1.10×10−6; figure 4B–D). Additionally, a comparison of the clusters and age at biopsy revealed a low average age of patients with samples in cluster 6, who shared an early onset feature; in that cluster, satellite cell components were over-represented (p=1.9×0−3; Welch’s t-test), and genes annotated with mitotic cell cycle ontology were altered (padj=1.29×10−7; figure 4B–D). Finally, three out of four control samples clustered into cluster 11 (figure 4C). We observed little evidence suggesting a correlation between causal genetic variants and clustering patterns, although the reconstruction of new NMF clusters using only cases in which a pathogenic variant was found resulted in a similar pattern to that from the entire cohort (online supplemental table 6; online supplemental figure 3). While cases without candidate genes were scattered across all clusters, cluster 10 was composed solely of such negative cases only (n=6). Notably, biopsy age and symptom onset age were highly correlated (R=0.95), ruling out the possibility that severe pathologies simply corresponded to biopsies performed later in disease progression (figure 4E). Nevertheless, a closer look at the differences in biopsy age and symptom onset age showed that the samples in cluster 8 presented the largest difference (9.45 years), whereas all other clusters showed negligible difference (online supplemental figure 4). This observation implies unique clinical features associated with cluster 8, including relatively slow progression and less visible symptoms. Cluster 6, in which biopsy was performed at age 0 in most samples, showed the lowest mean age difference (0 years). Therefore, the usage of muscle RNA-Seq data allowed the close correlation of histopathological findings, disease progression and genetic features.
Discussion
We provide evidence that a transcriptome-based platform can augment exome-based platforms for genetic diagnosis of NMD cases. Transcriptome analysis yielded a diagnostic ratio of 38.1% (75.0% from called variants and 25.0% from aberrant splicing events), surpassing that obtained via exome analysis (34.9%). Subsequent clustering analysis assisted in defining patients according to shared clinical features and gene expression signatures.
Several previous studies have used transcriptome data for variant calling but have not obtained satisfying concordances.22 23 Our approach was more successful as our candidate genes were highly expressed in muscles (online supplemental figure 5); additionally, improved bioinformatic tools have become available. Therefore, transcriptome-based variant calling and identification of aberrant transcripts can reproduce and augment exome-based variant calling in identifying disease-causing variants for patients with NMD.
Previous studies applying RNA-Seq to rare disease cohorts have employed aberrant splicing analysis to discover pathogenic transcripts in WES-negative patients, along with allele-specific expression and outlier gene expression analyses playing an auxiliary role (online supplemental table 7). In agreement with previous reports, we identified aberrant splicing events including exon skipping and intronic splice gains in various genes. While two cases were caused by variants at a canonical splice site, the other six were caused by deep intronic variants or deletions, which are difficult to detect by WES. WES read depth data are used to predict CNVs, but the exome capture and PCR amplification steps preclude accurate copy number estimation, leading to only limited use of WES data in calling pathogenic CNVs.24 Furthermore, despite the development of sophisticated computational tools for predicting effects of genetic variants on splicing, the synonymous genetic variant in LPIN1 and deep intronic variant in DMD were both incorrectly predicted from WES data. This observation further highlights the benefit of transcriptome sequencing in identifying pathogenic genetic variation.
The transcriptome-based approach provided additional utility in patient assessment through expression-based clustering (figure 4). NMF is a type of matrix factorisation algorithm that reveals low-dimensional structures from high-dimensional data such as gene expression data.25 26 It is widely used in transcriptomic data analysis, notably in cancer subtype profiling.27–29 Although our cohort was composed of patients with heterogeneous diagnoses and genetic causes, NMF indicated distinct molecular subtypes, suggesting common pathogenic pathways and potential targets for therapeutic intervention. For example, samples from patients with severe fibrosis and adipose replacement tended to cluster together, characterised by a distinctive transcriptomic pattern of altered lipid metabolism (cluster 8; figure 4). Patients subjected to muscle biopsy during infancy were shown to express cell cycle genes (cluster 6). Remarkably, these samples exhibited signatures of muscle regeneration, including the enrichment of mitotic cell cycle genes and markers of activated satellite cells.30 Since postnatal myofibres do not proliferate under normal circumstances,31 the enrichment of mitotic cell cycle genes in this cluster may imply active regeneration of muscle tissue or infantile muscle stem cell activity, which may help compensate for the muscle wasting.
Effective treatment options are not available for the majority of NMD cases due to their rarity and unique characteristics, complicated genetic pathophysiology and large portion of the affected area in the body.32 However, successful treatment strategies, such as antisense gene therapy in DMD, have recently been introduced in clinical practice. Eteplirsen is an Food and Drug Administration-approved ASO that induces the skipping of exon 51 in DMD to restore the reading frame.33 We applied a similar strategy of designing patient-specific ASOs that target splicing sites of CEs in two patients with DMD. We confirmed that the ASOs effectively induced the skipping of the CEs in patient myoblasts, suggesting a potential treatment options for these patients.
Additionally, the identification and targeting of specific cell types or biological pathways may be an option that can affect disease progression. For example, a study demonstrated that the inhibition of FAP adipogenesis delayed the progression of adipogenic muscle loss in limb girdle muscular dystrophy 2B model mice.34 Another study targeted satellite cells to improve muscle function and morphology in Pompe disease.35 Our clustering results identified a subset of patients with distinct genetic causes but common transcriptomic profiles, such as altered adipogenesis pathway alterations in FAP cells or abundant gene expression signatures of activated satellite cells and muscle regeneration, suggesting such pathways as potential therapeutic targets. Single-cell-level expression analysis would further enhance the power of our approach.
Histological, biochemical and ultrastructural analyses of affected muscle tissue are valuable for the precise diagnosis of patients with NMD. Thus, muscle biopsy is still an important diagnostic process and can also serve as a source of readily available diseased tissues. Relatively high tissue availability will facilitate transcriptomic analysis as a feasible diagnostic tool for NMDs. The limitations of RNA-Seq include sampling bias, namely, local tissue biopsy may not reflect overall physiological status of patient muscular system. Another disadvantage is that transcriptome analysis in the clinical setting requires a more complex tissue processing system, which includes extracting RNA from biopsied muscle tissue, sequencing and performing a more computationally intensive analysis. Genes with low muscle expression will be subjected to reduced variant sensitivity, but our analysis using GTEx data revealed that 94.7% (607/641) of NMD-associated genes and 74.8% (6001/8027) of the exons in those genes are expressed in the muscle (online supplemental figure 5).
Here we illustrate the value of transcriptome sequencing using biopsy samples for the diagnosis and genetic classification of patients with NMD. Transcriptome-based diagnosis can be used as an effective process to complement exome-based diagnosis for NMDs. In summary, our approach achieved (1) the efficient discovery of putative causal variants through variant calling, (2) the detection of aberrant transcripts and potential for patient-specific ASO application, and (3) the prediction of therapeutic effects through expression-based clustering, along with providing insights into the disease progression of NMDs.
Data availability statement
Data are available on reasonable request. Anonymised data not provided in the article may be shared at the request of any qualified investigator for purposes of replicating procedures and results.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Seoul National University Institutional Review Board (Nos 1707-126-872 and 1101-110-353). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank the patients and their parents who agreed to donate their tissues for this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
SEH and JK contributed equally.
Contributors Conceptualisation and funding acquisition: J-HC, MC. Data curation, software, formal analysis and visualisation: SEH, JaK. Investigation: SEH, JaK, SP, JL, SW, SK, SJ, JiK, MC. Resources: AC, MJK, J-SK, SYK, J-YS, J-HC. Supervision: J-HC, MC, J-YS. Writing–original draft: JaK, J-HC, MC. Writing–review and editing: SEH, JaK, JiK, J-YS, J-HC, MC. J-HC and MC are guarantors.
Funding This study was supported by the Korea Centers for Disease Control and Prevention (2021-ER0701-00), the Ministry of Health and Welfare (HI16C1986) and the National Research Foundation of Korea (2014M3C9A2064686 and 2020M3E5D7086836).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.