Article Text

Abstract
Background Among the several musculoskeletal manifestations in patients with Marfan syndrome, spinal deformity causes pain and respiratory impairment and is a great hindrance to patients’ daily activities. The present study elucidates the genetic risk factors for the development of severe scoliosis in patients with Marfan syndrome.
Methods We retrospectively evaluated 278 patients with pathogenic or likely pathogenic FBN1 variants. The patients were divided into those with (n=57) or without (n=221) severe scoliosis. Severe scoliosis was defined as (1) patients undergoing surgery before 50 years of age or (2) patients with a Cobb angle exceeding 50° before 50 years of age. The variants were classified as protein-truncating variants (PTVs), which included variants creating premature termination codons and inframe exon-skipping, or non-PTVs, based on their location and predicted amino acid alterations, and the effect of the FBN1 genotype on the development of severe scoliosis was examined. The impact of location of FBN1 variants on the development of severe scoliosis was also investigated.
Results Univariate and multivariate analyses revealed that female sex, PTVs of FBN1 and variants in the neonatal region (exons 25–33) were all independent significant predictive factors for the development of severe scoliosis. Furthermore, these factors were identified as predictors of progression of existing scoliosis into severe state.
Conclusions We elucidated the genetic risk factors for the development of severe scoliosis in patients with Marfan syndrome. Patients harbouring pathogenic FBN1 variants with these genetic risk factors should be monitored carefully for scoliosis progression.
- paediatrics
- genotype
- musculoskeletal diseases
- orthopaedics
Data availability statement
Data are available upon reasonable request. All genetic data included in this study are provided in online supplemental table 1. Other data are available on reasonable request. Deidentified data are available on reasonable request subject to ethics approval from the corresponding author.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Marfan syndrome (OMIM: #154700) was first described by Antoine Bernard Marfan in 1896 and is an autosomal dominant heritable disorder of the connective tissue.1 Marfan syndrome is characterised by several clinical manifestations, including dilatation of the aortic root, ectopia lentis and characteristic skeletal features. Among the several musculoskeletal manifestations in patients with Marfan syndrome, spinal deformity causes pain and restrictive ventilatory impairment and is a great hindrance to patients’ daily activities. Because patients with Marfan syndrome are potentially at high risk of further impairment of cardiopulmonary function following thoracotomy procedure and/or recurrent pneumothorax, it is essential to prevent the progression of spinal deformity and further deterioration of respiratory function. In addition, life expectancy of these patients has improved over the last few decades due to better medical and surgical management of cardiovascular conditions2; thus, appropriate control of spinal deformity is increasingly important. From the perspective of health economics, Marfan syndrome is reported to be the most common diagnosis among patients with syndromic scoliosis undergoing spinal deformity correction.3 Recently, we have reported that female sex and positive wrist signs are predictive factors for the progression of scoliosis in Marfan syndrome4; however, predicting the progression of spinal deformity is challenging, which leads to inadequate management of spinal deformity in patients with Marfan syndrome.
Up to 97% of patients with Marfan syndrome who fulfil the Ghent criteria have pathogenic variants in the FBN1 gene (OMIM: #134797), which contains 66 exons and encodes a major component of the extracellular matrix microfibril, namely fibrillin-1.1 5 More than 3000 pathogenic variants, which are mostly unique among families, have been identified in the FBN1 gene. The penetrance of FBN1 variants in Marfan syndrome is generally high, but phenotype prediction from these variants has been a challenging task. To date, several studies have demonstrated genotype–phenotype correlations in Marfan syndrome. Pathogenic variants in exons 25–33 of FBN1 were reported to be associated with neonatal Marfan syndrome,6–8 which is characterised by severe emphysema and mitral and/or tricuspid valve insufficiency in early childhood. Strong correlations between ectopia lentis and FBN1 variants affecting or creating cysteine residues have been repeatedly reported.9 10 Regarding aortic manifestations, some recent studies have shown that patients with haploinsufficient (HI) type FBN1 variants, such as nonsense and out-of-frame variants that presumably cause nonsense-mediated mRNA decay (NMD), have more severe aortic phenotypes than those with missense variants.11–15 However, there have been very few reports that investigated genotype–phenotype correlations between musculoskeletal manifestations and variant types of FBN1. Recently, Arnaud et al reported that the premature termination codon (PTC) variants in FBN1 are associated with the incidence of scoliosis with Cobb angle ≥20°.15 De Maio et al also found an association between stop codon variants in FBN1 and scoliosis with Cobb angle ≥20° or thoracolumbar kyphosis.16 However, no study has investigated the actual impact of the pathogenic FBN1 variant types on the progression of scoliosis into severe state requiring surgery. Scoliosis deteriorates patients’ quality of life when it progresses to a severe spinal curve, which causes worsening respiratory functions and/or low back pain.17 18 Hence, analyses focusing on patients with severe progressive spinal deformity are essential for eliciting clinically helpful information. The present study aimed to demonstrate, for the first time, the correlations between the pathogenic FBN1 variant types and the development of severe scoliosis to identify the genetic risk factors for progression of spinal deformity in patients with Marfan syndrome.
Methods
Patients and genetic tests
Data were retrospectively obtained from a prospective cohort from the Marfan syndrome center at our institute for a total of 175 months from September 2006 to March 2021. We enrolled consecutive patients with pathogenic or likely pathogenic FBN1 variants detected by genetic analysis. The variants were classified as pathogenic or likely pathogenic based on FBN1-specific variant classification guidelines,19 made by adapting 15 of the 28 general criteria of the American College of Medical Genetics and Genomics-Association for Molecular Pathology classification guidelines to better fit specific features of the FBN1 gene and Marfan syndrome. Identification of the pathogenic FBN1 variants was performed using Sanger sequencing for the FBN1 gene or next generation sequencing (NGS)-based genetic tests.14 NGS-based genetic tests included exome sequencing conducted using Japan’s Initiative on Rare and Undiagnosed Diseases in Pediatrics research and hybridisation capture-based gene panel testing for aortopathies conducted at the Kazusa DNA Research Institute (Chiba, Japan).14 20–22 In this study, seven patients with or suspected of having CNVs of the FBN1 gene were also included.22 Details of the genetic tests have been previously described.14 22 The reference sequence used for FBN1 was RefSeq NM_000138.4. Written informed consent was obtained either from the patients or from the guardians of minor patients.
Classification of pathogenic FBN1 variants
The pathogenic variants were classified into two main categories based on their location and predicted amino acid alterations: protein-truncating variants (PTVs) or non-PTVs. PTVs were defined as single nucleotide variants predicted to introduce a premature stop codon or to disrupt a splice site, small insertions or deletions predicted to disrupt a transcript’s reading frame, and larger deletions that remove the full protein coding sequence as previously described.23 Hence, in addition to PTC-creating variants, variants predicted to induce inframe exon-skipping (IFES) caused by disruptions of the splice donor site (eg, intron +1G or +2T) or splice acceptor site (eg, intron −1G or −2A) were included in PTVs. Out-of-frame and inframe exon-skipping variants detected by CNV analysis were also categorised as PTVs. Non-PTVs included missense variants and small inframe insertion or deletion variants, which are expected to exert dominant-negative effects.
Definition of severe and control groups
Patients with severe scoliosis were classified into the ‘severe’ group, which was defined as (1) patients who underwent primary surgery for scoliosis before 50 years of age or (2) patients with Cobb angle exceeding 50° before 50 years of age. This definition is used because major curves exceeding 50° progress even after skeletal maturity due to biomechanical reason,24 and thus in such patients prophylactic surgery is usually indicated to prevent progression of the curves to the severe level. Patients in the ‘control’ group were defined as those with a Cobb angle of 50° or less on the final X-ray which was taken at 15 years of age or older. To eliminate the impact of growth potential, which affects the progression of scoliosis, patients whose X-ray of the final follow-up was taken before 15 years of age were excluded from the control group. For all patients who did not undergo surgery for spinal deformity, posterior-anterior and lateral whole spine radiographs in standing position at final follow-up were evaluated and the Cobb angle was determined. For some patients who underwent surgery, the Cobb angle prior to surgery was unknown because preoperative X-rays were unavailable.
Statistical analysis
Fisher’s exact test was used to compare categorical data. Univariate and multivariate logistic regression analyses were performed to determine the risk factors associated with the progression of scoliosis to a severe state. Surgery-free curves were constructed using the Kaplan-Meier method and compared using the log-rank test. The threshold for significance was set at p<0.05. All statistical analyses were performed using JMP Pro (V.16.0.0; SAS Institute, Cary, North Carolina, USA).
Results
Demographic data of severe and control groups
Among 376 cases with pathogenic or likely pathogenic FBN1 variants, we identified a total of 278 eligible patients from 245 families, with 57 patients in the severe group and 221 patients in the control group. The remaining 98 cases were excluded for the following reasons: X-ray or clinical information was unavailable for 65 patients and final spinal X-ray was taken before 15 years of age in 33 patients. A total of 210 distinct FBN1 variants were identified in the 278 cases studied (online supplemental table 1). The details of the identified FBN1 variants are provided in online supplemental table 1. The profiles of the severe group are shown in table 1.
Supplemental material
Details of severe scoliosis cases with pathogenic FBN1 variants
There were 20 male and 37 female patients in the severe group. Among the 57 patients in the severe group, 42 underwent surgery, while 15 presented a Cobb angle exceeding 50° before 50 years of age (table 1). The mean age at first surgery for scoliosis in 42 patients was 15.2. All operations were identified as being performed for scoliosis or kyphoscoliosis. Demographic data comparing the severe and control groups are shown in table 2.
Demographic data of the severe scoliosis and control groups
The mean age at X-ray in the control group was 33.7 and the mean Cobb angle was 17.3° (table 2). Female sex and PTVs were identified more often in the severe group (table 2). When assessing PTVs, we observed a similar tendency towards increase in the frequency of PTC-creating variants and IFES variants in the severe group (table 2). Furthermore, when we constructed surgery-free curves using the Kaplan-Meier method, excluding 15 non-surgical severe cases, the equivalent impact of PTC-creating variants and IFES variants on the development of severe scoliosis was visually verified (online supplemental figure 1). These results confirmed the validity of our strategy of adopting the concept of PTVs,23 which included both PTC-creating variants and IFES variants. In contrast to the fact that PTVs were significantly more often identified in the severe group than non-PTVs, no significant difference was identified in the distribution of FBN1 variant types in patients with mild scoliosis (10° ≤ Cobb angle <30°) or moderate scoliosis (30° ≤ Cobb angle ≤50°) (figure 1 and table 2). This result suggested that FBN1 variant types may impact the progression rather than the onset of scoliosis (figure 1). Among the non-PTVs, there was no significant difference in the ratio of FBN1 variants affecting or creating cysteine residues between the two groups (table 2).
Supplemental material

Distribution of FBN1 variant types by severity of scoliosis. Significant difference in ratio for severe scoliosis was observed between the two variant types, while no significant difference in ratio for mild (10° ≤ Cobb angle <30°) or moderate (30° ≤ Cobb angle ≤50°) scoliosis was observed, suggesting that FBN1 variant type may affect the progression rather than the onset of scoliosis. PTV, protein-truncating variant. *P<0.05.
Distribution of pathogenic FBN1 variants
The detailed distribution of pathogenic FBN1 variants is shown in table 3 and figure 2.
Details of seven cases with exonic CNVs of FBN1
{kind=link}
{kind=link}
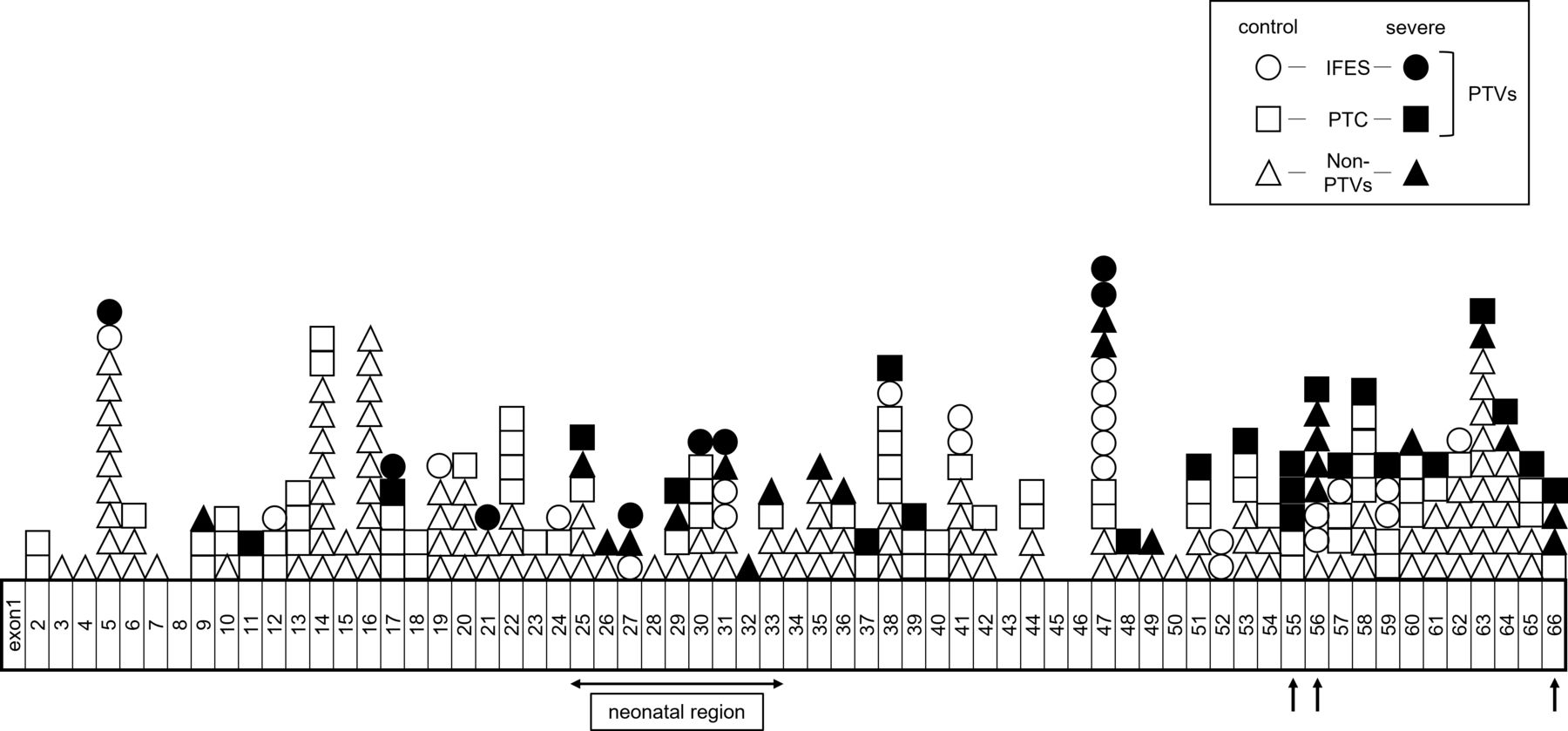
Detailed distribution of pathogenic FBN1 variants of 271 cases other than the CNV cases. Black arrows indicate possible hot spot regions for developing severe scoliosis. Control cases are depicted as white shapes, while cases in the severe group are depicted as black shapes. Circle, rectangle and triangle represent IFES, PTC-creating variants and non-PTVs, respectively. IFES, inframe exon-skipping; PTC, premature termination codon creating variants; PTV, protein-truncating variant.
Among the 278 cases with pathogenic or likely pathogenic FBN1 variants, 7 were suspected or confirmed to have CNVs22; 5 intragenic (multi-)exon deletions and 1 whole gene deletion were validated using chromosomal microarray analysis and multiplex ligation-dependent probe amplification analysis, respectively (table 3). In one patient (patient 330) clinically diagnosed with Marfan syndrome, heterozygous deletion of exons 64–66 was strongly suspected via a simple CNV prediction method that visually reviews the coverage tracks from the Integrative Genomics Viewer browser22 (table 3). The distribution of pathogenic FBN1 variants of the remaining 271 cases is summarised in figure 2.
Neonatal region (exons 25–33) as a hot spot for developing severe scoliosis
Because pathogenic variants in exons 25–33 of FBN1 were reported to be associated with early-onset severe cardiovascular phenotype in patients with Marfan syndrome,6–8 we hypothesised that there might be some ‘hot spot’ regions in the FBN1 gene for the development of severe scoliosis. First, we investigated whether the variants in this so-called ‘neonatal region’ (exons 25–33) were associated with severe scoliosis. Among the 34 cases with pathogenic FBN1 variants in this region, 13 (38.2%) developed severe scoliosis, while among the 244 cases with pathogenic FBN1 variants in other regions only 44 (18.0%) developed severe scoliosis. This was a significant difference (p=0.01) (figure 2 and table 3). This result indicates that harbouring pathogenic variants in the neonatal region might be associated with the development of severe scoliosis.
Univariate and multivariate analyses for identification of risk factors for developing severe scoliosis
To determine the actual impact of genetic factors on the development of severe scoliosis in Marfan syndrome, we conducted univariate and multivariate logistic regression analyses. Univariate analysis revealed that female sex (OR, 2.01; 95% CI 1.11 to 3.73), PTVs of FBN1 variants (OR, 2.03; 95% CI 1.13 to 3.73) and location of FBN1 variants in the neonatal region (OR, 2.81; 95% CI 1.28 to 6.00) all had a significant correlation with the development of severe scoliosis (table 4).
Univariate and multivariate logistic regression analyses for identifying the risk factors for the development of severe scoliosis in patients with pathogenic or likely pathogenic FBN1 variants
Multivariate analysis revealed that female sex (OR, 2.24; 95% CI 1.21 to 4.27), PTVs of FBN1 variants (OR, 2.30; 95% CI 1.25 to 4.33) and location of FBN1 variants in the neonatal region (OR, 3.11; 95% CI 1.38 to 6.88) were all significant independent predictive factors for the development of severe scoliosis in Marfan syndrome (table 4). To eliminate the impact of other genetic factors shared within the family members, we then conducted univariate and multivariate analyses for only 245 index cases as sensitivity analysis and achieved similar results (online supplemental tables 2 and 3). Moreover, to capture the time course impact of each risk factor on the development of severe scoliosis, we constructed surgery-free curves using the Kaplan-Meier method, excluding 15 non-surgical severe cases. The exclusion of the 15 non-surgical severe cases was necessary since the onset of ‘severe scoliosis’ with Cobb angle exceeding 50° could not be exactly determined due to lack of previous X-rays. Surgery-free curves constructed using the Kaplan-Meier method visually verified the similar impact of each risk factor on the development of severe scoliosis, although significance of the impact of PTVs on the development of severe scoliosis was marginal (online supplemental figure 2). This was probably due to the decreased number of severe cases. Furthermore, to capture the impact of the genetic factors more precisely, we constructed surgery-free curves using the Kaplan-Meier method for up to 20 years of age, excluding three cases who underwent surgery past the age of 20 who might have been modulated by other factors (online supplemental figure 3). Surgery-free curves up to 20 years of age visually verified the similar impact of each risk factor on the development of severe scoliosis as well (online supplemental figure 3).
Supplemental material
Impact of the genetic factors on the progression of existing spinal deformity
We then conducted another sensitivity analysis in which we limited the control cases with Cobb angle ≥10° to determine the impact of genetic risk factors on the progression of existing scoliosis. By eliminating the 58 cases without scoliosis (Cobb angle <10°), we obtained 57 severe and 163 control cases. The demographic data of this cohort are provided in online supplemental file 1. Univariate and multivariate logistic regression analyses demonstrated that female sex, PTVs of FBN1 variants and location of FBN1 variants in the neonatal region were all significant independent predictive factors for progression of scoliosis into severe state as well (online supplemental table 5).
Possible hot spot regions other than the neonatal region for developing severe scoliosis
Finally, we attempted to identify possible hot spot regions other than the neonatal region for severe scoliosis by visual inspection (figure 2 and table 3). We focused on the region where at least three cases developed severe scoliosis and more than 50% of the cases involved were categorised in the severe group. In this way, we identified exons 55–56 and the C-terminal region (exon 66) as possible hot spot regions (figure 2). Eight out of 14 cases (57.1%) harbouring pathogenic variants in exons 55–56 and 4 out of 5 cases (80.0%) with pathogenic variants in the C-terminal region (exon 66) developed severe scoliosis (figure 2 and table 3).
Discussion
This study provides two novel pieces of information. First, we demonstrated that the variant types of pathogenic FBN1 variants have distinct impacts on the progression of scoliosis in patients with Marfan syndrome. Second, we showed that not only variant types but also the location of FBN1 variants play an important role in the development of severe scoliosis.
There are several advantages in identifying patients at high risk of progression of spinal deformity. First, it can motivate patients at high risk of progression to seek regular medical help. Second, it may enable us to initiate timely interventions, including brace treatment and surgery. Because spinal deformity in Marfan syndrome is rapidly progressive and occasionally early onset, the timely initiation of brace treatment, which is usually suggested when the Cobb angle exceeds 20°, is sometimes difficult in these patients. This is probably one of the reasons for the lower success rate of brace treatment in Marfan syndrome than in idiopathic scoliosis.25–27 Regarding surgical management, surgery for spinal deformity in Marfan syndrome is reported to be accompanied by a higher incidence of complications compared with that in idiopathic conditions.28–30 Thus, it is crucially important to perform timely prophylactic surgery for scoliosis when the Cobb angle exceeds 45° or 50° to minimise perioperative complications, because surgery for progressed curves is known to be associated with a higher incidence of complications. Furthermore, impaired respiratory function due to progressive curves can be irreversible even after highly invasive surgery,31 especially when they have been left for a certain period. Finally, identifying patients at high risk of progression can be beneficial in terms of health economics.
The present study demonstrated that PTVs in FBN1 have distinct impacts on the development of severe scoliosis in patients with Marfan syndrome. In the current study, we adopted the concept of PTVs, which included PTC-creating variants and IFES variants. While most PTC-creating variants except those in the last exon result in haploinsufficiency through NMD mechanism, the actual functional effect of IFES remains to be determined. Furthermore, variants affecting splice sites in FBN1 very often result in IFES, since the number of the nucleotides in most of the exons (exons 4–63) in FBN1 is a multiple of 3. Thus, we first confirmed that PTC-creating variants and IFES variants have nearly equivalent impacts on the development of severe scoliosis. This finding is consistent with the findings of previous reports that demonstrated a significantly reduced amount of total mRNA of FBN1 in the splice variants, which was in agreement with a mechanism of haploinsufficiency.32 HI variants or PTC-creating variants in FBN1 have been proven to be associated with a higher risk of aortic events or aggressive aortic dilatation than dominant-negative variants.11–15 Hence, the present study proved that in Marfan syndrome, aortic manifestation and spinal deformity share common genetic risk factors for presenting severe phenotypes.
Pathogenic variants in the neonatal region (exons 25–33) have proven to be another genetic risk factor for developing severe scoliosis, regardless of the variant type (table 4). Faivre et al7 reported that pathogenic variants in this region are associated with the presence of scoliosis.7 However, no study has investigated the relationship between the severity of scoliosis and location of the FBN1 variants. Patients harbouring pathogenic variants in the neonatal region are known to exhibit variable severity of cardiovascular phenotypes and do not always present with neonatal Marfan syndrome, which is usually lethal in the first 2 years of life.7 8 33 Indeed, in the current case series of 13 cases with pathogenic FBN1 variants in the neonatal region in the severe group, 6 cases were free from cardiovascular surgery until the final follow-up (data not shown). Hence, it is important to recognise that patients with pathogenic variants in the neonatal region are at high risk of severe scoliosis because they do not always develop severe cardiovascular manifestations early in life and can be candidates for aggressive care for spinal deformity.
Exons 55–56 and the C-terminal region (exon 66) were also identified as possible hot spot regions for severe scoliosis. The reason for the correlation between severe patient phenotypes and these exon regions is unknown. Exons 55–56 encode two calcium-binding epidermal growth factor-like (cbEGF) domains: the cbEGF domains 34 and 35. Fibrillin-1 contains 43 cbEGF domains, each of which binds one calcium ion and is stabilised by three highly conserved disulfide bonds. Bound calcium stabilises cbEGF domains and cbEGF-cbEGF interdomain interfaces, extending tandem cbEGF domain repeats into rigid rod-like structures.34 Thus, variants in cbEGF domain are presumed to interfere with calcium binding, thereby perturbing microfibril assembly, structure and function,35 which may affect the structural strength of the spine. However, it remains to be elucidated why many patients in the severe group showed variations in this specific cbEGF region. The C-terminal region (exons 65–66) of FBN1 encodes asprosin, which is a novel glucogenic adipokine and is known to be involved in lipid metabolism.36 Furthermore, pathogenic variants in this region can cause Marfanoid-progeroid-lipodystrophy syndrome, a distinctive phenotype consisting of partial manifestations of Marfan syndrome, a progeroid facial appearance and clinical features of lipodystrophy.37 38 Because the association between idiopathic scoliosis and lower body fat has recently been suggested,39 variants in the C-terminal region of FBN1 may affect the progression of spinal deformity through altered lipid metabolism. In any case, further investigation is needed to determine whether pathogenic variants in these regions are associated with the development of severe scoliosis.
Recently, rare variants in FBN1 or FBN2 have been identified in patients with severe idiopathic scoliosis who did not clinically meet the diagnostic criteria for Marfan syndrome or Beals syndrome.40 Although there were not such cases in our present case series since at our institute genetic tests had been performed only in patients diagnosed as or highly suspected for Marfan syndrome with aortic or ocular manifestations according to the revised Ghent criteria,5 our present study may provide new insights into the aetiology of idiopathic scoliosis.
The present study identified female sex as another predictive factor for the progression of scoliosis in Marfan syndrome, which was consistent with our previous study.4 It has been repeatedly reported that male patients with Marfan syndrome have an increased risk of aortic events and root dilatation compared with female patients.13 14 41 42 This difference may partially explain the discrepancy in the severity between physical manifestation and aortic manifestation observed in individual patients with Marfan syndrome despite the common genetic risk factors.
In this study, we used a logistic regression model for multivariate statistical analysis. There were two reasons for selecting this model instead of the Cox regression analysis. First, the onset of ‘severe scoliosis’ with a Cobb angle exceeding 50° could not be determined for 15 non-surgical severe cases because consecutive previous spine X-rays were unavailable for these cases. Second, the proportional hazard assumption was not expected for progression of scoliosis in general because spinal deformity rapidly deteriorates during the growth spurt period and then progresses very slowly. Hence, surgery for scoliosis is usually performed prophylactically in late adolescence when the Cobb angle exceeds 45°–50°. In the current study, the mean age at first surgery for scoliosis in 42 cases was 15.2, and the Kaplan-Meier surgery-free curves for scoliosis clearly demonstrated that most operations had been performed during adolescence (table 1 and online supplemental figures 1 and 2).
This study had a few limitations. First, it was retrospective in design. Second, there might be some selection bias. Due to the lack of whole spine X-rays, not all patients with pathogenic FBN1 variants were analysed in this study. Third, in some patients undergoing surgery, the preoperative severity of spinal deformity is unknown due to the lack of preoperative X-rays. Finally, the effect of pathogenic variants on the stability and function of the protein product was not verified.
To the best of our knowledge, this study, for the first time, determined the genetic risk factors for progression to severe spinal deformity in patients with Marfan syndrome. PTVs in FBN1 have distinct impacts on the development of severe scoliosis in patients with Marfan syndrome. Variants in the neonatal region were also independent genetic risk factors for the development of severe scoliosis. Exons 55–56 and the C-terminal region (exon 66) in FBN1 were also identified as possible hot spot regions. Therefore, patients harbouring pathogenic FBN1 variants with these genetic risk factors should be monitored carefully for scoliosis progression.
Data availability statement
Data are available upon reasonable request. All genetic data included in this study are provided in online supplemental table 1. Other data are available on reasonable request. Deidentified data are available on reasonable request subject to ethics approval from the corresponding author.
Ethics statements
Patient consent for publication
Ethics approval
This study was approved by the Research Ethics Committee of the University of Tokyo (#2674-(4), G1538-(11)).
Acknowledgments
We would like to thank Editage (https://www.editage.jp/https://www.editage.jp/) for editing and reviewing this manuscript for English language.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Conception and design: YT, NT, RI. Acquisition of data: YT, YM, SK, HYag. Formal analysis: RI, TD. Interpretation of data: YT, HYam, YO. Methodology: YT, RI, MA. Project administration: NT, YO. Supervision: ST. Writing - original draft: YT. Writing - review and editing: NT and RI. Guarantor: YT
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.