Article Text

Abstract
Background Musculocontractural Ehlers−Danlos syndrome is caused by biallelic loss-of-function variants in CHST14 (mcEDS-CHST14) or DSE (mcEDS-DSE). Although 48 patients in 33 families with mcEDS-CHST14 have been reported, the spectrum of pathogenic variants, accurate prevalence of various manifestations and detailed natural history have not been systematically investigated.
Methods We collected detailed and comprehensive clinical and molecular information regarding previously reported and newly identified patients with mcEDS-CHST14 through international collaborations.
Results Sixty-six patients in 48 families (33 males/females; 0–59 years), including 18 newly reported patients, were evaluated. Japanese was the predominant ethnicity (27 families), associated with three recurrent variants. No apparent genotype–phenotype correlation was noted. Specific craniofacial (large fontanelle with delayed closure, downslanting palpebral fissures and hypertelorism), skeletal (characteristic finger morphologies, joint hypermobility, multiple congenital contractures, progressive talipes deformities and recurrent joint dislocation), cutaneous (hyperextensibility, fine/acrogeria-like/wrinkling palmar creases and bruisability) and ocular (refractive errors) features were observed in most patients (>90%). Large subcutaneous haematomas, constipation, cryptorchidism, hypotonia and motor developmental delay were also common (>80%). Median ages at the initial episode of dislocation or large subcutaneous haematoma were both 6 years. Nine patients died; their median age was 12 years. Several features, including joint and skin characteristics (hypermobility/extensibility and fragility), were significantly more frequent in patients with mcEDS-CHST14 than in eight reported patients with mcEDS-DSE.
Conclusion This first international collaborative study of mcEDS-CHST14 demonstrated that the subtype represents a multisystem disorder with unique set of clinical phenotypes consisting of multiple malformations and progressive fragility-related manifestations; these require lifelong, multidisciplinary healthcare approaches.
- musculoskeletal diseases
- human genetics
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Ehlers−Danlos syndrome (EDS) is a clinically and genetically heterogeneous group of heritable connective tissue disorders characterised by skin hyperextensibility, joint hypermobility and tissue fragility.1 2 Musculocontractural EDS (mcEDS) is a rare type of EDS, caused by biallelic loss-of-function variants either in the gene for carbohydrate sulfotransferase 14 (CHST14)/dermatan 4-O-sulfotransferase 1 (D4ST1)(CHST14, mcEDS-CHST14) (MIM#601776) or in the gene for dermatan sulfate epimerase (DSE) (DSE, mcEDS-DSE) (MIM#615539).2 3 D4ST1 and DSE are critical enzymes for biosynthesis of dermatan sulfate (DS), which is covalently attached to specific serine residue of core protein to form glycosaminoglycan (GAG) chains for proteoglycan, in the Golgi apparatus. DSE catalyses epimerisation of the C-5 position of glucuronic acid (GlcA) residues in a chondroitin precursor backbone, resulting in the formation of the dermatan backbone. Subsequently, D4ST1 transfers a sulfate group from 3′-phosphoadenosine-5′-phosphosulfate to the C-4 hydroxy group of N-acetylgalactosamine adjacent to iduronic acid (IdoA) in dermatan backbone, resulting in the formation of mature DS.1 3
Dysfunction of D4ST1 or DSE causes a depletion of DS, completely replaced by chondroitin sulfate (CS) in affected skin fibroblasts, and especially in the GAG chains of decorin, a major DS-proteoglycan that contributes to collagen fibril assembly.4–7 The ultrastructure of skin samples from patients with mcEDS-CHST14 included dispersed dermal collagen fibrils, resulting from the conformationally linear CS chains stretching from the outer surface of collagen fibrils to adjacent fibrils; in contrast to regularly and tightly assembled collagen fibrils of healthy control samples, according to DS chains that were round and wrapping collagen fibrils.5 8 This spatial disorganisation of collagen networks is suggested to disrupt the ring-mesh structure of GAG side chains surrounding collagen fibrils and result in marked skin hyperextensibility and fragility.8 9
mcEDS-CHST14 was originally regarded as three independent conditions: a rare type of arthrogryposis syndrome (adducted thumb-clubfoot syndrome)4; a specific type of EDS (EDS, Kosho type)5 10 and a subset of kyphoscoliosis type without lysyl hydroxylase deficiency.6 These conditions were later recognised to represent a single clinical entity with highly characteristic features comprising multiple malformations (eg, craniofacial features, multiple congenital contractures, ocular and visceral malformations) and progressive fragility-related manifestations (eg, skin hyperextensibility and fragility, joint hypermobility with luxation, progressive spinal and foot deformities, large subcutaneous haematomas, visceral ruptures).11 Clinical features that correspond to the characteristics of EDS, along with the presence of altered collagen biosynthesis, justify its designation as a type of EDS.11
Major diagnostic criteria for mcEDS-CHST14 are (1) multiple congenital contractures, characterised by adduction–flexion contractures and talipes equinovarus; (2) characteristic craniofacial features, evident at birth or in early infancy; and (3) characteristic cutaneous features including hyperextensibility, bruisability and fragility with atrophic scars and increased fine palmar creases.2 Thus far, 48 patients in 33 families have been reported,4–6 10 12–27 including six individuals from five families whose spinal manifestations were described.27 However, accurate prevalences of each feature/complication and detailed natural history data remain unknown.
We further delineated the spectrum of pathogenic variants, prevalences of various manifestation and natural history of mcEDS-CHST14, using detailed and systematically collected clinical and molecular findings of 48 reported and 18 newly identified patients with diverse demographic characteristics.
Methods
The cohort included previously described patients and newly identified patients, diagnosed through direct sequencing of CHST14 or a next-generation sequencing-based custom panel analysis for heritable connective tissue disorders,28 using DNA from peripheral blood lymphocytes (online supplemental information S1). Primer sequences for CHST14 are available on request. Clinical information included detailed and comprehensive issues, and issues relating to the course of the disorder (eg, perinatal courses, life span, causes of death, episodes of dislocations and large subcutaneous haematomas). An Excel file-based questionnaires to collect the clinical information were filled out by international contributing physicians of 38 patients. Such clinical information was directly collected through medical records from 13 patients who underwent medical check-ups in Shinshu University Hospital. Clinical information was also collected through reviewing literatures on 47 patients. All collected information were integrated into a large dataset. The cumulative incidences of the initial joint dislocations and the first episodes of large subcutaneous haematomas in patients with mcEDS-CHST14 were analysed using EZR statistical software (V.1.38).29
Clinical data of eight reported patients with mcEDS-DSE7 22 30 31 were collected through literatures and questionnaires (four patients) from referring physicians to compare with mcEDS-CHST14. The non-parametric Mann-Whitney U test was used for calculating the differences between the prevalence of common clinical features of patients with both subtypes, using EZR statistical software (V.1.38).29
Facial images of patients with mcEDS-CHST14 were analysed using the DeepGestalt technology available in Face2Gene Research application (FDNA, USA). In order to evaluate our impression that these patients alter their facial appearances between childhood and adolescence−adulthood, we performed comparative facial analysis of childhood (younger than 8 years old) and adolescence−adulthood (8 years old or older). We selected 15 patients, whose facial photographs are available both in their childhood and their adolescence−adulthood. Composite images were generated for each period, and binary comparison between these images was measured based on the area under the receiver operating characteristic curve (AUC).32 An AUC value of 1.0 indicates complete separation between facial features in their childhood and in their adolescence−adulthood, while an AUC value of 0.5 indicates no discrimination between these cohorts.32
Results
Sixty-six patients (33 male patients, 33 female patients) in 48 families were included in this study. Eighteen patients in 15 families were newly identified in this study, including a male infant whose brother was previously described.18 Of the 66 patients, 65 were diagnosed with mcEDS-CHST14 based on both clinical evaluation and molecular confirmation of pathogenic variants in CHST14. Only one reported patient was diagnosed with mcEDS-CHST14 based on clinical evaluation in the context of the family history without molecular diagnosis because he died immediately after birth. Common clinical features are summarised in table 1; clinical issues (physical features, events, complications and treatment) described in a single patient are listed in online supplemental table S1; detailed molecular data are shown in online supplemental table S2; and clinical photographs are shown in figures 2–4. Median patient age on collection of clinical information was 19 years (n=63; range <1 day to 59 years). Ethnic origins varied, but were concentrated in Asia: Japanese in 27 families (56%), followed by Turkish and Hispanic (n=3 each); Indian, Dutch and ‘Asian’ (n=2 each); and Austrian, Afghani, Miccosukee, Curaçaoan, Moroccan, Pakistani, Mexican, Thai/German and Bangladeshi (n=1 each).
Supplemental material
Supplemental material
Summary of clinical features in 66 patients with mcEDS-CHST14, compared with eight reported patients with mcEDS-DSE
Genotypes
Pathogenic variants in CHST14 are outlined in figure 1. The results of major in silico analyses and large-scaled population database search of each variant are shown in online supplemental table S2. The reference protein is NP_569735.1; the reference mRNA sequence is NM_130468.3. Twenty-six intragenic variants were identified, including 13 missense, seven frameshift (deletions, duplications and deletion–insertion), five nonsense (one base substitution, deletion and duplication) and one in-frame deletion. Most missense variants were located in the middle of the sulfotransferase 2 domain; truncating (frameshift or nonsense) variants were located outside of the sulfotransferase 2 domain or on both ends of the domain (figure 1A). Recurrent variants included P281L in 20 Japanese families (42%), F209S in 10 Japanese families (21%), Y293C in five Japanese families (10%), V49* in three families (Turkish, Dutch), R213P in two families (Austrian, Hispanic), R218L in two families (Dutch, Thai/German-Irish) and E262K in two families (Hispanic, Mexican). Five novel variants were included (p.L137P, p.W162*, p.R218L, p.I241K, p.R320*). Based on the 2015 guideline by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology,33 two novel nonsense variants (p.W162* and p.R320*) and three novel missense variants (p.L137P, p.R218L and p.I241K) were classified as ‘pathogenic’ and ‘likely pathogenic’, respectively (online supplemental table S2).
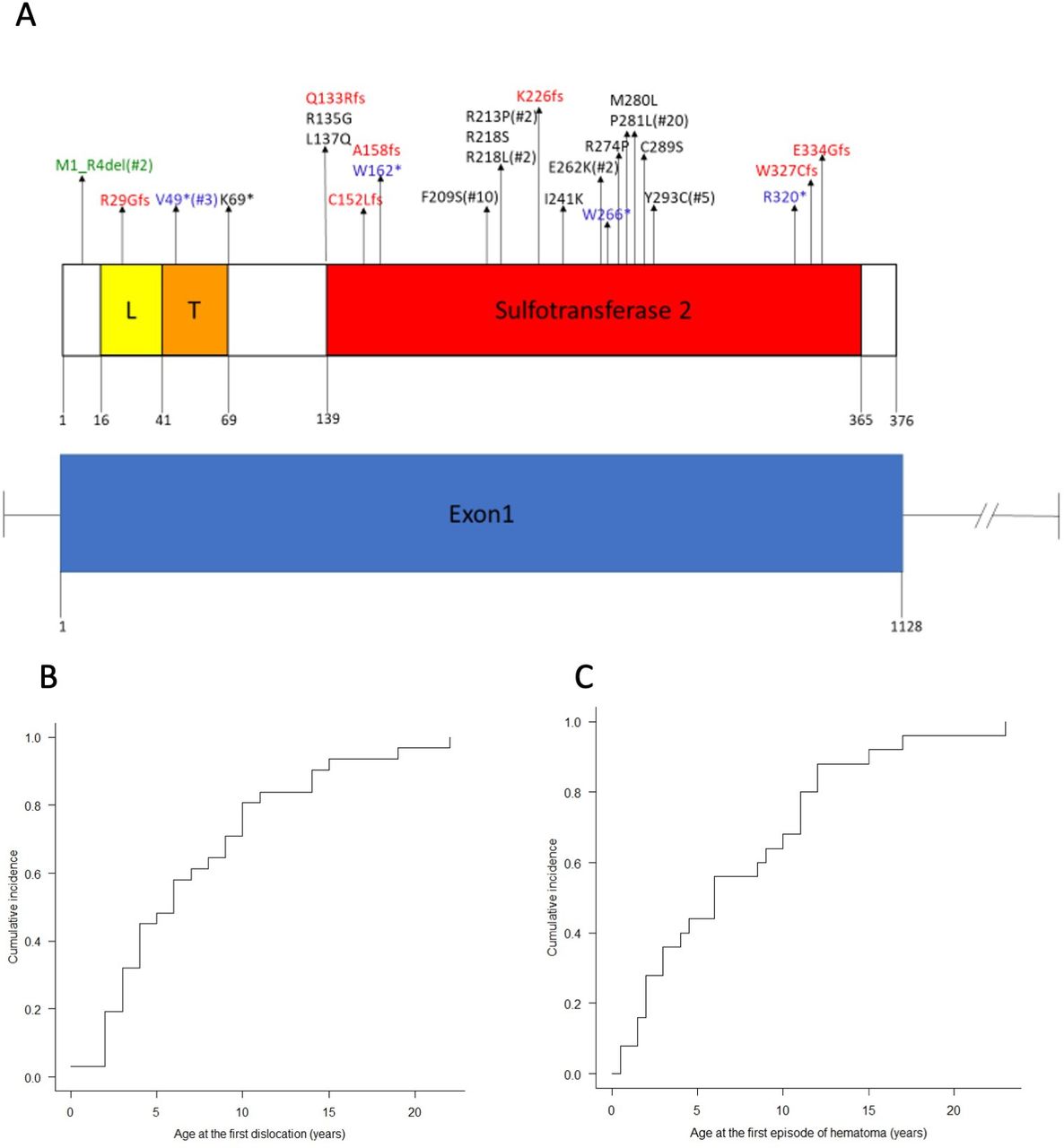
(A) Distribution of CHST14 variants in 48 families with mcEDS-CHST14. CHST14 genomic structure and CHST14 protein domains were constructed by reference to NM_130468.3 and NP_569735.1 using Pfam (http://pfam.xfam.org), respectively. CHST14 is a single-exon gene that comprises a low complexity domain (L), a transmembrane domain (T) and a sulfotransferase 2 domain. Arrows indicate the location of each variant. Recurrent variants are marked with *#, which represents the number of families with that variant. (B) Cumulative incidence of the first episode of dislocation. (C) Cumulative incidence of the first episode of developing large cutaneous haematoma. Most patients experienced the first incidence of dislocation or large subcutaneous haematoma before late childhood.
Perinatal history
Pregnancies were complicated in 36% (15/42): reduced fetal movement (n=4); hand and/or foot abnormalities and obstetric problems (n=3 each); intrauterine growth impairment, oligohydramnios, hydronephrosis and brain abnormalities (n=2 each) and single umbilical artery (n=1). Deliveries were eventful in 34% (16/47): caesarean section (n=11) due to breech presentation (n=5) and fetal distress (n=3) and others (n=1 each, online supplemental table S1). Neonatal periods were eventful in 89% (42/47): congenital contractures (n=27), including talipes equinovarus in 20 and adducted thumbs in six; poor suckling or feeding (n=13), including tube feeding in five; hypotonia or muscle weakness (n=8); cardiopulmonary problems (eg, apnoea, cyanosis, respiratory distress, persistent pulmonary hypertension, repeated cardiac arrest) (n=5); hyperbilirubinaemia, cryptorchidism with/without inguinal hernia and asphyxia (n=3 each); hydronephrosis, ophthalmologic problems (eg, congenital cataract, retinal haemorrhage) and intracranial problems (eg, intracranial calcification, ventricular enlargement) (n=2 each) and others (n=1 each, online supplemental table S1).
Life span
Nine deceased patients were included; their median survival was 12 years (range <1 day to 59 years). Cause of death was available for five patients: respiratory distress shortly after birth, septicaemia due to scalp haemorrhage at age 12 years, infective endocarditis at 28 years, colonic perforation at 37 years and intracerebral haemorrhage at 59 years.
Craniofacial features
Merged images of facial photographs (figure 2A), generated through Face2Gene application, as well as representative photographs (figure 2C) are shown in figure 2. Craniofacial features were distinctive, and some features became less evident or changed with age: a round facial shape with micro-retrognathia in early childhood, which became slender and elongated with a protruding jaw, crowded teeth and facial asymmetry in adolescence or adulthood. Difference between facial features in their childhood and in their adolescence−adulthood were calculated as statistically significant with an AUC value as 0.904 (p=0.027) (figure 2B). Other features included characteristic forehead (n=12; broad, bossed and flat); dental problems (n=8; multiple caries, discoloured teeth, malocclusion and tooth defect), bushy eyebrows with/without synophrys (n=5); broad nose or nasal bridge (n=4); pointed chin, downturned corners of mouth, drooping lower eyelids or face, prominent or flat occiput, hypoplastic supraorbital ridges, ptosis and malar hypoplasia (n=3 each), proptosis and protruding eyes (n=2) and others (n=1 each, online supplemental table S1).

Craniofacial features. (A) Composite images of 15 patients with mcEDS-CHST14 in their childhood patients versus in their adolescence−adulthood using the Face2Gene facial recognition technology. (B) The binary comparison using Face2Gene demonstrates a difference between the facial features of childhood and adolescence to adulthood (area under the curve (AUC)=0.945; p=0.016 and receiver operating characteristic: (ROC)). (C) Clinical photographs of craniofacial features. A blue square indicates a single patient. Regardless of age or sex, most patients share common craniofacial characteristics, including hypertelorism, downslanting and short palpebral fissures, short nose with hypoplastic columella and long philtrum. Several features changed from childhood to adolescence: facial shape changed from round to slender and elongated, while microretrognathia changed to a protruding jaw.
Skeletal features
Skeletal features were also distinctive (figures 3 and 4). In addition to multiple congenital contractures, characteristic finger morphologies included tapering (n=32), slender (n=25), cylindrical (n=22), long (n=20), camptodactyly/clinodactyly/overlapping fingers (n=7), finger-like thumbs and spatulate (n=2 each) and arachnodactyly and short (n=1 each). Progressive talipes deformities included planus (n=26), valgus (n=23), equinovarus/varus (n=8), cavus (n=6), broad (n=4), small/short and hallux valgus (n=3 each) and rockerbottom feet and metatarsus adductus (n=1 each). Hypermobile joints were limited to small or finger joints in 12 patients; and were observed at larger, both small and larger joints, or generalised in 24. Recurrent dislocation occurred in shoulder(s) (n=28), knee(s) (n=20), elbow(s) or hip(s) (n=14 each), patella(e) (n=8), radial head(s) (n=7), radioulnar joint(s) (n=6), finger(s) (n=3) and temporomandibular joint, ankle, wrist, glenohumeral joint, metacarpophalangeal joint or toes (n=1 each). Median age at the initial dislocation was 6 years (n=32; range, 0–22 years) and the calculated cumulative incidence showed that 80% of patients with joint dislocations had their initial episode by the age of 10 (figure 1B). Spinal deformities included scoliosis (n=24; mild in 11, moderate in three, and severe in four), kyphoscoliosis (n=11; severe in eight) and kyphosis (n=6; mild in one and severe in one). Pectus deformities included flat or shield (n=17), excavatum (n=13), thin or narrow (n=10), flat (n=17), carinatum/protrusion (n=5), asymmetric (n=4) and round (n=1). Other skeletal features included finger abnormalities (n=7; eg, ulnar deviation, narrowing of interphalangeal spaces, short fourth metacarpal, syndactyly), injuries (n=5; fractures of tibial plateau, Salter-type 2 fracture of the proximal phalanx of the second toe, elbow fracture, knee tendon and meniscus), vertebral abnormalities (n=5; tall vertebrae; narrow, high, and osteophytic vertebral bodies; non-rib bearing lumbar type vertebral bodies and cervical spondylosis), rib abnormalities (n=3; thin ribs, 11 pairs of ribs), toe abnormalities (n=2; interphalangeal contractures, overlapping toes), pain (n=2; chronic articular pain) and others (n=1 each, online supplemental table S1). Congenital contractures of fingers became milder with age, although some impairment of finger extension and/or flexion persisted.

Clinical photographs of hands. A blue square, a red square and a green square indicate a finger shape, joint mobility of fingers and wrists and palmar creases of a single patient, respectively. Patients display typical hand-related features: adducted thumbs; characteristic finger shapes (eg, slender, tapering and/or cylindrical); variable extension or flexion contracture of distal interphalangeal joints, proximal interphalangeal joints and metacarpophalangeal joints; joint hypermobility of fingers and wrists; and acrogeria-like fine palmar creases.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clinical photographs of feet, thorax, skin, teeth and large subcutaneous haematoma. A blue square, a red square, a green square, a purple square and a yellow square indicate a foot shape, a pectus shape, skin features, teeth and a large subcutaneous haematoma of a single patient, respectively. Patients display talipes deformities including congenital equinovarus and progressive planus and/or valgus with characteristic toes (eg, long, slender and/or deformed). Patients display flat chest or pectus excavatum. Skin involvement includes hyperextensibility, fragility with atrophic scars and fistula formation. Mild to severe tooth irregularity, a high and narrow palate and a large subcutaneous haematoma in the thigh are shown.
Cutaneous features
Cutaneous features associated with hyperextensibility and fragility were common (figures 3 and 4). Hyperalgesia to pressure was also frequent (74%), which typically hindered blood pressure measurement in the upper arms. Recurrent subcutaneous infections occurred occasionally (37%), typically accompanied by fistula formation around the buttocks (n=9) or elbows (n=3). Other skin features included widely spaced nipples (n=8); soft, smooth, doughy and/or velvety skin (n=8); thin and/or translucent skin (n=6); single transverse palmar creases, hirsutism, finger nail abnormalities (small, double-layered), molluscoid pseudotumors, clavus of the sole and hyperkeratosis (elbows, upper leg) (n=2 each) and others (n=1 each, online supplemental table S1).
Cardiovascular features
Large subcutaneous haematoma was the most common cardiovascular complication (figure 4), occurring in the scalp (n=25), buttocks (n=14), thighs (n=9), lower leg (n=8), leg (n=7), knee (n=6); face, chest, sacral region, extremities or foot (n=2 each) and arm, elbow, back, spine or ankle (n=1 each). Median age at the initial episode of haematoma was 6 years (n=25; range 0–23 years) and the calculated cumulative incidence showed that 80% of patients with large subcutaneous haematomas had their initial episode by the age of 12 (figure 1C). Cardiac valve abnormalities included mitral regurgitation (n=13; trivial in four and mild in one), mitral prolapse (n=10), aortic regurgitation (n=2) and tricuspid regurgitation (n=1). Congenital heart defects (n=11) included atrial septal defects (n=9) and coarctation of the aorta, patent ductus arteriosus, dextrocardia and tricuspid atresia (n=1 each). Mild enlargement of ascending aorta was detected in one patient (3%). Other cardiovascular or related complications included prolonged bleeding time (n=4) and others (n=1 each, online supplemental table S1).
Pulmonary and gastrointestinal features
Pneumothorax initially occurred at ages 23, 28 and 31 years in three patients; it was accompanied by a large bulla in one patient. Bilateral pleural effusion was observed in one. Constipation was the most common gastrointestinal complication. Diverticula, mostly in the colon (n=6) or small intestine or stomach (n=1 each), resulted in perforation in four patients at ages 12, 24, 24 and 29 years. Other gastrointestinal complications included abdominal pain (eg, colic, cramping), diarrhoea (n=4 each), a frog croak-like large bowel sound (n=3), gastric ulcer (n=2) and others (n=1 each, online supplemental table S1).
Genitourinary features and sexual development-related findings
Cryptorchidism in male patients was the most common urogenital abnormality. Hydronephrosis was associated with ureteropelvic junction obstruction in three patients. Bladder dysfunction presented as dilatation or urinary retention (n=8) or neurogenic bladder (n=2). Genitourinary abnormalities in a single case are listed in online supplemental table S1. Sexual development-related findings included poor breast development and hypogonadism. Median age of menarche was 14 years (n=10; range 12–16 years).
Ophthalmologic and otological features
The most common ophthalmologic complications were refractive errors including myopia (n=33), astigmatism (n=12), hyperopia (n=6) and amblyopia (n=1). Median age at retinal detachment was 15.5 years (n=6; range 9–20 years). Blindness was noted in six patients, associated with glaucoma, macular bleeding, lacquer crack in Bruch’s membrane or retinal detachment. Other ophthalmologic abnormalities included microcornea (n=18), other corneal abnormalities (circumferential pannus, pigmentation, opacification/vascularisation, irregular, plana and sclerocornea) (n=6), fundus abnormalities (peau d’orange fundus, papilledema/retinal haemorrhage/retinal atrophy/pale papilla and congenital optic nerve atrophy), iris abnormalities (peripheral/posterior anterior synechia and atrophic), amblyopia and cataract (n=3 each); microphthalmia, shallow anterior chamber, angioid streaks and scleromalacia (n=2 each) and others (n=1 each, online supplemental table S1).
Twenty-two patients exhibited hearing loss; high-frequency loss was more common (n=10) than low-frequency loss (n=2). Other otologic abnormalities included otitis media (n=5) and others (n=1 each, online supplemental table S1).
Neuromuscular and neurodevelopmental features
Recurrent neurological, muscular and neurodevelopmental manifestations included structural brain anomalies, tethered spinal cord and muscular hypotonia accompanied by motor developmental delay. Structural brain abnormalities, observed in 61% (20/33), included ventricular abnormalities (n=15; enlargement in 12 patients, asymmetry in two and both in one), aplasia or hypoplasia of septum pellucidum (n=5), hypoplastic corpus callosum (n=3), Dandy-Walker anomaly and cerebellar hypoplasia (n=2 each) and others (n=1 each, online supplemental table S1). Neurological features included hyperactive tendon reflexes and seizures (n=2 each) and others (n=1 each, online supplemental table S1). Serum creatine kinase level was elevated in 50% (n=7/14; 277, 698 and 807 U/L in females; 573, 1065, 1838 and 336–2944 U/L in males; range 41–153 in females and 59–248 in males). Other muscular features included muscle weakness and/or poor muscle mass (n=7), diastasis recti (n=4), fatigability and myopathic findings on conduction study or histology (n=2 each) and others (n=1 each) (online supplemental table S1). Median age on walking independently was 26 months (n=39): 1 year (n=3), 13–23 months (n=12), 24–35 months (n=8), 36–47 months (n=6), 48–60 months (n=5), 5 years (n=1), 6 years (n=2), 7 years (n=1) and 15 years (n=1). One adult patient could not walk unsupported because of orthopaedic problems. No significant intellectual disability was noted in patients of school age or older.
Treatment
Surgical intervention was performed, planned or considered in 96% of patients (45/47) for various complications: talipes equinovarus (n=28), retinal detachment (n=11), cryptorchidism (n=8); large subcutaneous haematoma, spinal deformity (kyphoscoliosis and scoliosis) and dislocation (hip, patella, cervical spine and elbow) (n=7 each), inguinal hernia (n=6); contracture (adducted thumbs and camptodactyly), diverticular perforation/diverticulitis, glaucoma and urologic disorders (hydronephrosis) (n=4 each), cardiac disorders (atrial septal defect and mitral valve prolapse) and umbilical hernia (n=3 each); foot deformity, gastric ulcer, (hemo)pneumothorax, knee abnormalities and synovitis/bursitis (n=2 each) and others (n=1 each, online supplemental table S1). The median number of complications requiring surgical intervention per patient was 3 (n=43; range 1–8). Braces or orthotics were required in 80% of patients (28/35) for congenital or progressive skeletal complications: talipes equinovarus (n=20), foot deformities (n=6), hand-wrist deformities and kyphoscoliosis (n=4 each), hip dislocation (n=3) and meniscus injury (n=1). Various pharmacological therapies were administered in 70% of patients (28/40): laxatives or related gastrointestinal drugs for constipation (n=12), 1-desamino-8-D-arginine vasopressin (DDAVP) for prevention and treatment of large subcutaneous haematoma (n=6), eye drops for prevention/treatment of glaucoma (n=5), medication for osteopenia/osteoporosis (n=4), cardiovascular medication (n=3); analgesics, prophylactic antibiotics for postoperative spinal fistula or urinary tract infection and iron supplementation (n=2 each) and others (n=1 each, online supplemental table S1). Additionally, 76% of patients (22/29) underwent rehabilitation: physiotherapy alone (n=15) or physiotherapy with occupational therapy (n=7).
Comparison with mcEDS-DSE
Detailed clinical information of eight reported patients with mcEDS-DSE was combined in the integrated dataset along with patients with mcEDS-CHST14. Common clinical features and complications were added in table 1. Although clinical features were typically shared by both subtypes, statistical analyses showed that the frequencies of low-set ears, joint manifestations (hypermobility and recurrent joint dislocation), skin features (hyperextensibility, bruisability, fragility and atrophic scars), constipation and refractive errors were significantly higher (p<0.01) in patients with mcEDS-CHST14 than in those with mcEDS-DSE (table 1).
Discussion
This study represents the first international and the largest cohort of patients with mcEDS-CHST14. It provides a detailed and comprehensive catalogue of clinical and molecular features associated with mcEDS-CHST14. A complete list of pathogenic variants in CHST14 from various ethnic backgrounds, accurate prevalence of multisystem manifestations and clinical courses of important complications are described.
Characteristic craniofacial, skeletal and cutaneous features were shared by all patients and could be recognisable during the neonatal period or infancy. Distinctive craniofacial and skeletal features evolved gradually from early childhood to adolescence or adulthood as mentioned in previous case series10 17 18; and, regarding craniofacial features, we have created merged images of facial photographs in their childhood as well as in their adolescence−adulthood (figure 2A) and statistical significance was shown between the two periods (figure 2B). Talipes equinovarus, treated with braces or surgery, resulted in various types of progressive deformities. Congenital contractures of fingers tended to become milder and peripheral joint hypermobility tended to become prominent. Spinal deformities typically included mild scoliosis and occasionally progressive thoracolumbar kyphosis, accompanied by reduced physiological thoracic spine curvature, cervical kyphosis and vertebral malformations.27 Cutaneous features, as described previously and common in other types of EDS,10 17 18 included hyperextensibility, bruisability and fragility in all. Fine palmar creases without normal major palmar creases or flexion creases of fingers (figure 3) would suggest mcEDS-CHST14.3 10 17 All these major features in the disorder support the validity of minimal diagnostic criteria proposed in the 2017 International Classification: congenital multiple contractures and characteristic craniofacial features at birth or in early infancy; congenital multiple contractures and characteristic cutaneous features in adolescence and in adulthood.2
Thirteen missense variants, seven frameshift variants, five nonsense variants and one in-frame deletion were listed in this cohort. No apparent genotype–phenotype correlation was noted, presumably because truncating variants and missense variants would both result in complete loss-of-function of D4ST1; this was demonstrated in recombinant cells or patients’ skin fibroblasts (for P281L, C289S and Y293C).5 Three highly recurrent variants (c.842C>T, p. P281L; c.626T>C, p.F209S; c.878A>G, p.Y293C), detected only in Japanese patients and registered in multiple population database with their allele frequencies (online supplemental table S2), occurred presumably as a result of founder effects. Predominance of the disorder in Japanese population was attributable to these recurrent variants.
In the perinatal period, affected fetuses could exhibit characteristic ultrasonographic findings including adducted thumbs and talipes equinovarus; breech presentation and/or fetal distress could occur, requiring caesarean delivery. Affected neonates typically have multiple congenital contractures (eg, adducted thumbs and talipes equinovarus) and hypotonia with/without poor suckling. Skeletal complications constitute a major physical problem in childhood. Talipes equinovarus, treated with braces with/without surgery, resulted in progressive deformities that may hinder walking unsupported or motor development. Spinal deformities are common and could require treatment with braces or surgery in severe cases. Joint dislocations generally occur by the age 10 years (figure 1B). Another major physical concern in childhood is large subcutaneous haematomas, which typically occur by the age of 12 years. Constipation, recurrent urinary tract infections with/without bladder dysfunction, hearing loss, refractive error, glaucoma, retinal detachment and dental problems are also important, with onset in childhood. From adolescence to adulthood, female patients generally show poor breast development. Reproductive function of affected individuals remains unknown; pregnancy/delivery among female patients or partners of affected male patients has not been described so far. Pneumothorax, colonic diverticula, gastric ulcer and cardiac valve abnormalities are rare but serious complications that could influence morbidity and mortality.
Healthcare guidelines for mcEDS-CHST14 would be proposed as follows, based on the current study and previous literatures.3 18 34 Typically, the diagnosis could be suspected in the neonatal period or early infancy from characteristic craniofacial and skeletal features. The differential diagnosis includes Loeys–Dietz syndrome, Freeman–Sheldon syndrome, spondylodysplastic EDS and kyphoscoliotic EDS.3 Molecular confirmation involves Sanger sequencing of CHST14 and DSE, or a next-generation sequencing-based panel analysis of relevant genes.2 28 Additional approaches include copy number detection to identify large deletions in these genes and a urinary disaccharide analysis of CS and DS chains to demonstrate the absence of DS moiety.35 On diagnosis, systemic surveillance is necessary to detect multiple congenital abnormalities or complications (eg, systemic radiographic and ultrasonographic examinations, ophthalmological and otologic screening, brain and spine MRI). With respect to progressive fragility-related multisystem manifestations, regular evaluations (orthopaedic, cardiovascular, urologic, ophthalmologic, otologic and dental) are recommended from childhood. When patients ambulate independently, special attention should be paid to gait instability due to progressive talipes deformities and muscle hypotonia, which could result in injury-related joint dislocations, skin laceration and large subcutaneous haematoma. Foot orthosis is recommended for talipes deformities and early intervention (physiotherapy) is recommended for motor developmental delay associated with muscle hypotonia. Intranasal DDAVP therapy would be a reasonable choice to prevent large subcutaneous haematoma.10 17 In adolescence, close orthopaedic examinations are required for progressive spinal deformities. Psychosocial support could be considered for adaptation to disability and enhancement of self-esteem from childhood or adolescence. Regarding limitations in school activities and future employment, consultations with teachers or employment specialists might be helpful. In adulthood, regular systemic evaluations should be continued and genetic counselling, including discussions regarding reproduction, could also be considered. Management of acute serious complications (eg, dislocations, skin laceration, large subcutaneous haematoma, diverticulitis with/without perforation, pneumothorax, retinal detachment) in coordination with emergency room, intensive care unit and relevant departments would be crucial and a specific emergency card could be useful.
This is the first attempt to evaluate clinical difference statistically between mcEDS-CHST14 and mcEDS-DSE. Core skin features (hyperextensibility, bruisability, fragility and atrophic scars) were significantly more frequent in mcEDS-CHST14. An ultrastructural study of skin samples from patients with mcEDS-CHST14 demonstrated that dermal collagen fibrils were dispersed, which were caused by the conformationally linear CS chains stretching from the outer surface of collagen fibrils to adjacent fibrils.8 On the other hand, collagen fibrils of healthy control samples were regularly and tightly assembled, which were associated with DS chains that were round and wrapping collagen fibrils.8 This spatial disorganisation of collagen networks, presumably related to a complete compositional change from DS to CS in GAG chains, was suggested to disrupt the ring-mesh structure of GAG side chains surrounding collagen fibrils and result in marked skin hyperextensibility and fragility.8 9 36 The mcEDS-DSE represents milder phenotypes compared with mcEDS-CHST14, and partially synthesised DS in the affected skin fibroblasts might be related to the residual DSE activity or compensation by DSE2 encoded by DSE-like (DSEL) that is a homologue of DSE.7 Since the number of reported patients with mcEDS-DSE is small and longitudinal data are limited at present, more accurate and comprehensive phenotypic comparison between mcEDS-CHST14 and mcEDS-DSE remains to be clarified.
Inevitable limitation could exist as a retrospective survey on an extremely rare disorder, including a possible incomplete dataset of features and complications based on the absence of a surveillance guideline as well as a possibility of recall bias especially for features and complications in the early childhood of older patients. However, this study would present the most accurate, detailed and comprehensive clinical information that could be obtained at present through a multiple data collection strategy (questionnaires, medical records and literatures) based on a unified clinical information file.
In conclusion, this first international collaborative study offers further delineation of clinical and molecular features in mcEDS-CHST14. Unique, multisystemic clinical manifestations, more severe than those for mcEDS-DSE, require lifelong, multidisciplinary healthcare. The pathophysiology of this syndrome is gradually being elucidated, and future aetiology-based therapies such as adeno-associated virus-based gene therapy and chaperone therapy might be expected.36
Supplemental material
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study was approved by the Ethics Committee at Shinshu University School of Medicine (Matsumoto, Japan) (610, 628, 5080) and Yokohama City University Graduate School of Medicine (Yokohama, Japan) (A110929007).
Acknowledgments
We are grateful to the patients and their families and the EDS Society (https://www.ehlers-danlos.com) for cooperation during this study. We thank Dr Cecilia Giunta (Division of Metabolism, Connective Tissue Unit, University Children’s Hospital Zurich, Zurich, Switzerland), Dr Sohei Watanabe (Department of Orthopaedics, Nakajima Hospital, Sendai, Japan) and Dr Takeshi Yagyu (Department of Cardiovascular Medicine, National Cerebral and Cardiovascular Centre, Suita, Japan) to help collect information of the patients. We also thank Dr Shuji Mizumoto, PhD (Department of Pathobiochemistry, Faculty of Pharmacy, Meijo University, Nagoya, Japan) for critical advice on glycobiology, Nicole Fleicher (FDNA Inc, USA) for a collaborative analysis using the Face2Gene RESEACH and Ryan Chastain-Gross, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors MM and TK interpreted the data and drafted the manuscript. TK conceived the work and organized the data collection. AU assisted with data collection. TY, N Matsumoto and N Miyake conducted the molecular study. The others followed up on patients and provided data. All authors participated in revision and approval of the manuscript.
Funding This study was supported by Research on Intractable Diseases (073) (2012–3) (TK) and Research Programme on Policy of Measures for Intractable/Rare Diseases (20FC1046) (2020–2) (TK), Ministry of Health, Labour and Welfare, Japan; Practical Research Project for Rare/Intractable Diseases (105) (2015–7) (TK), Programme for an Integrated Database of Clinical and Genomic Information (16kk0205001h0501, 16kk0205012h1001) (2016–2020) (TK) and (JP19ek0109280, JP19dm0107090, JP19ek0109301, JP19ek0109348, and JP18kk020501) (N Matsumoto) and the Initiative on Rare and Undiagnosed Diseases (IRUD) (19ek0109301h0002) (2018–2020) (TK), Japan Agency for Medical Research and Development (AMED); Grant-in-Aid for Scientific Research (C) (25460405) (2013–5) (TK), Grant-in-Aid for Scientific Research (B) (19H03616) (2019–2021) (TK) and (JP19H03621) (N Miyake), from the Japan Society for the Promotion of Science, Japan; Medical Research Encouragement Prize of the Japan Medical Association (2013) (TK); Japan Foundation for Pediatric Research (2014) (TK); Problem-Solving Oriented Training Programme for Advanced Medical Personnel: NGSD (Next Generation Super Doctor) Project (TK) and the Intramural Research Programme of the National Human Genome Research Institute. Division of Clinical Sequencing, Shinshu University School of Medicine, is an endowment division, supported with an unrestricted grant from BML Inc. and Life Technologies Japan Ltd. Delfien Syx and Fransiska Malfait are research fellows at the Research Foundation Flanders.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.