Article Text

Abstract
Background Fetal akinesia (FA) results in variable clinical presentations and has been associated with more than 166 different disease loci. However, the underlying molecular cause remains unclear in many individuals. We aimed to further define the set of genes involved.
Methods We performed in-depth clinical characterisation and exome sequencing on a cohort of 23 FA index cases sharing arthrogryposis as a common feature.
Results We identified likely pathogenic or pathogenic variants in 12 different established disease genes explaining the disease phenotype in 13 index cases and report 12 novel variants. In the unsolved families, a search for recessive-type variants affecting the same gene was performed; and in five affected fetuses of two unrelated families, a homozygous loss-of-function variant in the kinesin family member 21A gene (KIF21A) was found.
Conclusion Our study underlines the broad locus heterogeneity of FA with well-established and atypical genotype–phenotype associations. We describe KIF21A as a new factor implicated in the pathogenesis of severe neurogenic FA sequence with arthrogryposis of multiple joints, pulmonary hypoplasia and facial dysmorphisms. This hypothesis is further corroborated by a recent report on overlapping phenotypes observed in Kif21a null piglets.
- nervous system diseases
- neuromuscular diseases
Data availability statement
Data are available upon reasonable request. All variants have been deposited into ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under Institute of Medical Genetics and Applied Genomics, University of Tübingen, including VCV… through VCV….
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Fetal akinesia (FA) comprises a clinically heterogeneous group of disorders characterised by absent or reduced fetal movements resulting in a variety of secondary deformations.1 Associated clinical presentations range in severity from distal arthrogryposis, multiple pterygium syndrome and arthrogryposis multiplex congenita (AMC) to severe FA deformation sequence (FADS) or lethal congenital contracture syndrome.2 Observed clinical features might overlap between these entities and depend on the degree and time point of the movement impairment during pregnancy. Consistent findings include joint contractures, reduced skeletal muscle mass, subcutaneous oedema, fetal hydrops, pulmonary hypoplasia, intrauterine growth restriction and craniofacial dysmorphisms.3 4 Additional organ systems might be involved as part of a broad syndromic spectrum associated with the primary cause of FA. While many of these conditions are recognised in utero, others are not detected until birth.5
The causes of FA are diverse and include maternal or environmental causes as well as a rapidly growing number of genetic factors with at least 166 distinct loci being firmly associated with FA to date.6 Although individually rare, they collectively account for an incidence of multiple joint contractures in 1/2000–1/5000 live births.7 8
Significant progress has been made over the last years to define the set of genes involved and substantially improve the diagnostic yield.6 9 10 However, despite the wide availability of exome sequencing even in a prenatal routine diagnostic setting, a relevant portion of affected individuals and their families remain without a firm diagnosis. This situation complicates genetic counselling of families regarding prognosis and risk of recurrence and prevents the development of targeted therapeutic approaches.
We report the results of an exome sequencing study in 23 index cases prenatally or perinatally diagnosed with FA. In addition to the diagnostic evaluation of known disease genes, an extended analysis to prioritise potential new disease genes was performed in a scientific context.
Materials and methods
Study cohort
Written informed consent was obtained from all probands or legal guardians. The affected individuals and their families were recruited in routine clinical care at different sites. Apart from family 22, which has been investigated at the genetikum Stuttgart, all other biosamples were submitted for exome-based prenatal or postnatal diagnostic testing to the Institute of Medical Genetics and Applied Genomics (Tübingen). All individuals had intrauterine or perinatal clinical presentations suggestive of FA and shared arthrogryposis with contractures of the joints in at least two different body parts as a common feature.4 In-depth phenotyping included evaluation of prenatal fetal organ abnormalities, whenever possible the clinical findings from follow-up examinations or the autopsy documentation from fetal pathologist in prenatally lethal FA or terminated pregnancies. Data and clinical reports were provided by the primary specialised healthcare provider of the patients.
Genetic studies
Exome sequencing was conducted on genomic DNA of at least one affected proband per family. Coding genomic regions were enriched using a SureSelect XT Human All Exon Kit V.6 or V.7 (Agilent Technologies, Santa Clara, California, USA) for subsequent sequencing as 2×125 or 2×100 bp paired-end reads on a HiSeq2500 or NovaSeq6000 system (Illumina, San Diego, California, USA). Generated sequences were analysed using the megSAP pipeline (https://github.com/imgag/megSAP). Clinical variant prioritisation included different filtering steps (eg, minor allele frequency (MAF) <0.1% in 1000 g, ExAC or gnomAD (https://gnomad.broadinstitute.org; assessed June 2021), and an in-house database) and was conducted independently by two trained diagnostic molecular geneticists according to an in-house standard operating procedure. Trio exome analyses were performed as part of a fast track process established at the institute for prenatal investigations and paediatric intensive care patients. For these cases, a final diagnostic report was achieved within 9–23 days with a mean turnaround time of 14.7±4.8 days (median 15 days).
In patient F22:II.4, a Human Core Exome kit (Twist Bioscience, South San Francisco, California, USA) was used for enrichment of coding sequences and generated libraries were sequenced on a NextSeq 500 platform (Illumina, San Diego, California, USA). Generated bam files were transferred for secondary evaluation using the megSAP pipeline.
Subsequent variant validation and carrier testing of additional family members was performed by Sanger sequencing. Primer sequences and PCR conditions are available upon request.
Prioritised variants were classified following the recommendations of the American College of Medical Genetics and Genomics.11 Affected individuals carrying likely pathogenic or pathogenic clinically relevant variants were considered to have a firm diagnosis. Patients harbouring a likely pathogenic variant compound heterozygous with a variant of uncertain significance were considered to be potentially solved.
For candidate disease gene prioritisation, a cohort analysis of the eight unsolved index cases was performed to identify potentially pathogenic variants in genes affected in more than one index case.
Results
Phenotypical FA spectrum
Twenty-three index patients diagnosed with FA were recruited as part of the study. In addition, 5 further affected siblings were examined for a total of 28 patients. Of these, 14 were male and 14 were female. In 17 cases, the pregnancy was terminated, the child was stillborn or died shortly after birth. One patient died at the age of 2.5 months. Ten patients were alive with ages ranging from 2 months to 9 years. Documented phenotypical features are summarised in tables 1 and 2 as well as in online supplemental table S1.
Supplemental material
List of genetic variants and clinical details of individuals with a likely diagnosis
Identified KIF21A (NM_001173464.2) variants and associated clinical features
Diagnostic yield in FA
In 13 of 23 (56%) of the index cases, the disease-causing variants were detected in 12 different known FA-associated genes. Apart from TTN, which was observed in two families, all other gene defects were identified only once (table 1). We observed a total of 17 unique variants of which 12 have not been reported previously. In 7 of 13 (54%) of the individuals with genetically confirmed FA, the pattern of inheritance was compatible with autosomal recessive inheritance. Of these, two of seven (29%) of the cases carried homozygous variants. A hemizygous change in X-chromosomal GPC3 was inherited from a healthy heterozygous carrier mother. In 3 of 13 (23%) of the firm diagnoses de novo, dominant variants were identified as the likely cause of the disease. Of note, two heterozygous changes were maternally inherited with the mother being similarly affected (F9) or having a milder disease presentation associated with a mosaic state of the variant (F1). Two of 23 (9%) of the index cases remained unsolved with variants of uncertain significance in the OMIM FA disease genes SLC6A9 and BICD2 (a detailed discussion of the phenotypical and molecular findings is provided in the online supplemental data). In 8 of 23 (35%) of the cases, no suggestive variants in OMIM disease genes were prioritised and the molecular cause remained unclear in a diagnostic context.
Supplemental material
Bi-allelic KIF21A loss-of-function variants in FA
In the eight unsolved index patients, an extended analysis was performed to investigate putative novel gene–disease associations. Pathogenic variants in known FA genes were absent in these eight families. DNA variant lists of the eight unsolved index cases were jointly investigated using strict filters for allele frequency, function and conservation. KIF21A was identified in a search for genes shared across these FA families carrying rare bi-allelic variants. The required filters were non-synonymous homozygous or putatively compound heterozygous variants, rare in gnomAD (MAF <0.001), and predicted to cause functional damage (combined annotation dependent depletion, >20). This filtering strategy left two index cases, individuals F22:II.4 and F23:II.1, carrying different homozygous predicted loss-of-function variants in KIF21A (NM_001173464.2): the stop variant c.1346T>A, (p.Leu449*) in exon 9 and the frameshift variant c.2371del, (p.Arg791Glufs*8) in exon 17 (figure 1B). The mutant transcripts are likely to be degraded by nonsense-mediated mRNA decay. Both changes were not observed in gnomAD and an in-house database containing exome and genome datasets of >15 000 individuals with unrelated phenotypes. No other bi-allelic protein-truncating variants (PTVs) in KIF21A were observed in these databases. Sanger sequencing confirmed a homozygous state of the familial KIF21A variants in all affected fetuses with the parents and a healthy sibling being heterozygous carriers.

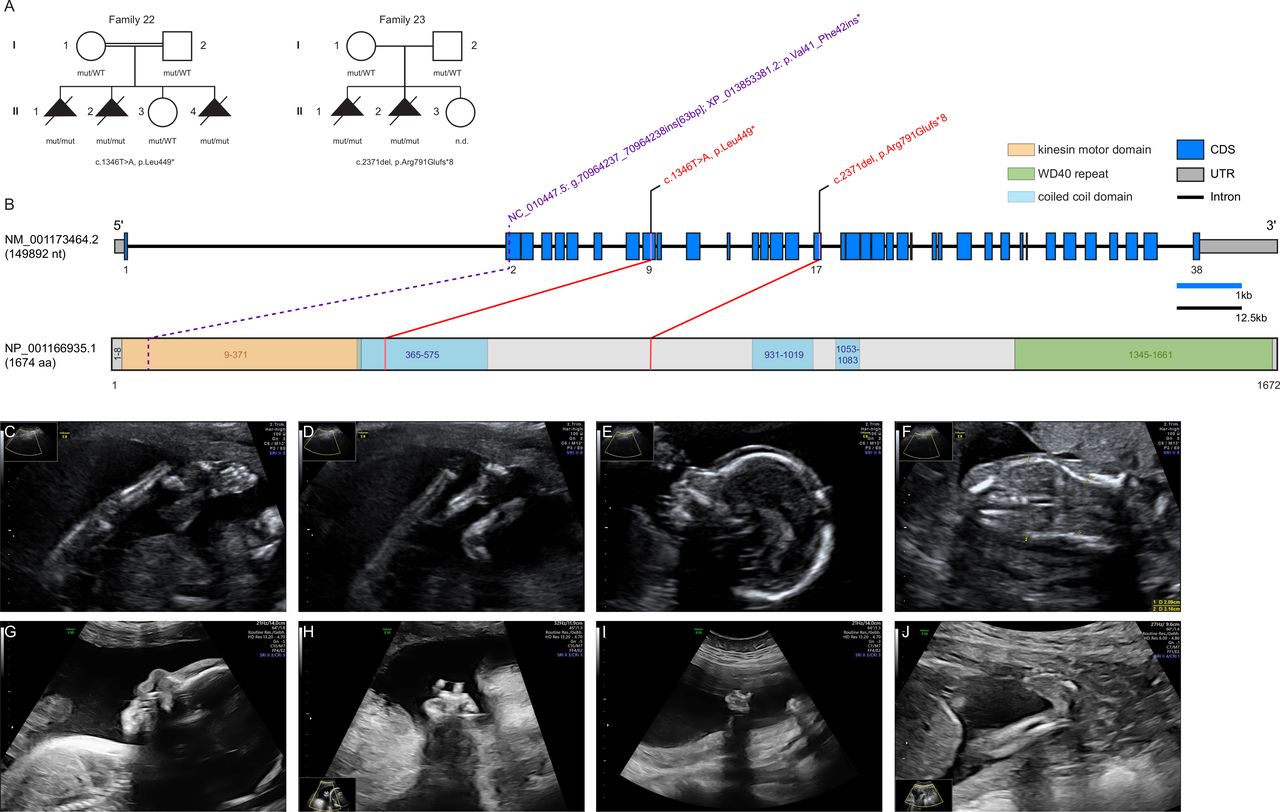{kind=link}
Pedigrees of investigated KIF21A families, structure of KIF21A and prenatal ultrasound scans. (A) Pedigrees of two families with pathogenic variants in KIF21A, illustrating the variant carrier status of affected (closed symbols) and healthy (open symbols) family members. Identified variants are provided below the pedigrees. (B) Gene structure of KIF21A with known protein domains and motifs of the gene product and localisation of the identified variants (red). The change reported in the Kif21a orthologue in Sus scrofa is shown in purple. Intronic regions are not drawn to scale (black scale bar indicates 12.5 kb of intronic regions, blue scale bar indicates 1 kb of exonic regions). (C–F) Representative ultrasound scans of individual F22:II.4 at 19+0 weeks of gestation: (C) lower extremity with persistently extended and crossed legs with clubfeet; (D) both knee joints fixed in extension; (E) micrognathia and slight prenasal oedema; (F) coronal ultrasound through the thorax and abdomen shows thoracic hypoplasia. (G,H) Ultrasound scans of individual F23:II.1 at 25+5 weeks of gestation: (G) facial profile with micrognathia and prenasal oedema; (H) right hand with crossed fingers. (I,J) Ultrasound scans of individual F23:II.2 at 21+0 weeks of gestation: (I) left clenched hand; (J) lower extremity with clubfoot. CDS, coding sequence; mut, mutated allele; n.d., not determined; UTR, untranslated region; WT, wildtype allele.
Phenotypes associated with loss of KIF21A
The clinical features observed in a total of five similarly affected KIF21A-mutant fetuses from two families are summarised in table 2 and listed in detail in online supplemental table S2. Pedigrees of families F22 and F23 as well as representative ultrasonography findings are provided in figure 1A and C–J. Both families originate from Turkey and consanguinity was reported for parents of family F22.
Supplemental material
In all fetuses, first abnormalities were recognised by high-resolution fetal ultrasonography between the 19th and 26th week of gestation. In five of five fetuses, reduced fetal movements were documented associated with multiple joint contractures. These included clenched fingers (four of five), fixed extension or flexion of the wrists (three of five), elbows (two of five) and knees (four of five) as well as talipes equinovarus (five of five). Marked thoracic hypoplasia was observed in five of five fetuses and polyhydramnios in four of five fetuses. Facial dysmorphisms were micrognathia (four of five) and/or retrognathia (four of five) combined with inconsistent variable additional features including brachycephaly (one of five), hypertelorism (one of five, autopsy), low-set ears (two of five, autopsy), flat broad nose (one of five, autopsy), scalp oedema (four of five), and prenasal and/or neck oedema (two of five). Skeletal deformations were documented in ultrasonography or autopsy in two of five fetuses with thoracic kyphoscoliosis, straight ribs and slender tubular bones. Other ultrasonographic or autopsy findings included gastrointestinal abnormalities (three of five) such as diaphragmatic hernia with protrusions (two of five) as well as cerebral ventriculomegaly (one of five), dextrocardia (one of five), clinodactyly (one of five) and urogenital abnormalities with bilateral tortuous ureters (one of five). All pregnancies were terminated between the 21st and 29th week of gestation. The ultrasonographic findings were mostly congruent with documentations of autopsies conducted on fetuses F22:II.1, F22:II.2 and F23:II.1. The lung weight to body weight ratio was determined in individuals F22:II.1 (0.009; 23+1 weeks of gestation; normal >0.015) and F23:II.1 (0.009; 28+5 weeks of gestation; normal >0.012) and suggested severe lung hypoplasia.
Together, these molecular and phenotypical data are in line with bi-allelic KIF21A PTVs being implicated in the pathogenesis of a severe form of autosomal recessive FA characterised by arthrogryposis multiplex, pulmonary hypoplasia and variable facial dysmorphisms.
Discussion
The aim of this study was to further define the molecular bases underlying FA using exome sequencing. Our results confirm the diagnostic yield and broad genetic heterogeneity in FA reported in previous studies including myogenic, neuromuscular junction, neurogenic as well as syndromic malformation aetiologies.6 Individuals with less severe prenatal findings surviving the first months and years of life were largely diagnosed with myopathies or syndromic malformations. On the molecular level, our findings underline the importance of variant-specific or transcript-specific mechanisms and genotype–phenotype correlations.6 9 Illustrative examples include the identification of variants in GPC3 (OMIM *300037) and TTN (OMIM *188840), two genes mostly known for their association with Simpson-Golabi-Behmel syndrome (OMIM #312870) and (cardio-)myopathies (OMIM #604145, #613765, #608807, #603689, #611705), respectively. The GPC3 variant c.1666G>C, (p.Gly556Arg) observed in patient F3:II:1 alters the same residue affected by different variants [c.1667G>T, (p.Gly556Val) and c.1666G>A, (p.Gly556Arg)] detected in patients with Simpson-Golabi-Behmel syndrome. Of note, the latter one predicts the same alteration on amino acid level.12–15 This residue is located in a region critical for GPC3 cleavage during the process of plasma membrane anchoring. Functional studies demonstrated for the change c.1666G>A, (p.Gly556Arg), that the protein was not glycanated and stayed in the cytoplasm instead of being attached to the cell surface.12
In two unrelated individuals, the recurrent nearsplice variant c.39 974–11T>G, (p.?) affecting the metatranscript of TTN (NM_001267550.2) was observed in compound heterozygosity with two different loss-of-function alleles. This change has been recently reported in 10 individuals from 8 families diagnosed with AMC and is postulated to alter the expression of a TTN isoform predominantly expressed during fetal skeletal muscle development.16 17 This observation exemplifies the possibility that transcript-specific alterations may cause distinct clinical presentations via interference with early prenatal and postnatal processes.
Noteworthy, our study provides further evidence of two loci being associated with FA, namely NEK9 and KIF21A. NEK9 encodes a serine-threonine protein kinase that interacts with the chromatin structure modulating FACT (FAcilitates Chromatin Transcription) complex and is essential for interphase progression.18 In addition, NEK9 plays a role in regulating chromosome alignment and segregation during mitosis.19 Casey et al first associated a homozygous nonsense variant in NEK9 c.1489C>T, (p.Arg497*) with lethal skeletal dysplasia in two Irish traveller families. Their results suggested nonsense-mediated decay of mutant NEK9 mRNA to result in delayed cell cycle progression and reduced proliferation. Furthermore, patient-derived fibroblasts exhibited a defect in ciliary function.20 However, to our knowledge, only one additional individual with FADS harbouring another homozygous PTV variant [c.1498del, p.(Glu500Lysfs*33)] has been published thereafter.21 Our report on compound heterozygous predicted loss-of-function variants in an additional similarly affected unrelated proband provides important additional evidence of bi-allelic NEK9 variants to be associated with FA.
Apart from methodological advantages to detect certain types of genetic variation, a key advantage of exome and full genome sequencing over panel-based approaches targeting a predefined set of genes is the perspective to establish new disease genes. A combined filtering strategy tailored to prioritise new factors associated with FA led us to identify bi-allelic PTVs in the kinesin family member 21A gene (KIF21A), a member of the kinesin-4 family, in several affected fetuses from two families. Kinesins (KIFs) are molecular motor proteins that constitute 15 kinesin families with 44 members in the human genome. They directionally transport multiple cargos such as organelles, protein complexes, vesicles, mRNA or virus particles along microtubule tracks, using ATP to drive conformational changes that generate motile force.22 Owing to their molecular function, they play an essential role in various cellular processes, including cell-cycle dynamics and progression, ciliogenesis and cilia function as well as organisation of polar cells during organogenesis.23 Pathogenic variants in several kinesin superfamily proteins (KIF5C, KIF14, KIF26B) have been associated with different disease entities, including FA.24–26
KIF21A is highly conserved in bilateria with, for example, 96.23% identity of the amino acid sequence in Sus scrofa (online supplemental data) and 54.07% in Caenorhabditis elegans. It is highly enriched in axons, dendrites and muscle, and inhibits microtubule growth at the cell cortex.27–29 Gain-of-function variants in KIF21A have been associated with autosomal-dominant congenital fibrosis of extraocular muscles (CFEOM1, OMIM #135700). The protein consists of an amino terminal motor domain, central stalk domain and carboxy terminal domain containing WD40 repeats.30 In its inactive state, the third coiled-coil stalk domain binds the motor domain, inhibiting its interaction with microtubules. When KIF21A is activated, this motor domain is released and can bind to microtubules.30 Of note, all variants associated with CFEOM1 so far are clustering in the third coiled-coil stalk and motor domain, suggesting that these changes disrupt interaction of the two domains, leaving KIF21A in a constitutively active state.30
Supplemental material
To our knowledge, recessive-type variants compatible with a loss of KIF21A function have so far not been associated with clinical phenotypes in human. However, Fang et al identified a 63 bp insertion in exon 2 of the porcine Kif21a gene predicted to result in a truncated protein lacking the complete motor domain.31 32 Piglets with a homozygous insertion were either stillborn or died shortly after birth with congenital malformations resembling arthrogryposis multiplex congenita. Of note, eye movements of heterozygous pigs have not been investigated in this study.31
The extreme genetic heterogeneity underlying FA translates into a broad spectrum of biological functions of affected proteins and disease mechanisms. These include ion channels or pumps, receptors or modulators, inborn errors of metabolism, factors involved in transcription and translation, cell cycle, cell signalling/secretion as well as motor proteins or protein trafficking.6 From an organelle perspective, a postulated mechanism underlying FA is impaired ciliogenesis,33 which is interesting, as KIFs are known to fulfil an essential role in ciliogenesis and cilia function.23 In addition, primary cilia-driven signalling regulates growth cone dynamics and axonal tract development,34 and several genes linked to arthrogryposis, such as AUTS2, CBL, DNM2, IGHMBP2, KIF5C, SETX, SNAP25 and TOR1A function in growth cone regulation.35 It has been demonstrated that relief of KIF21A autoinhibition causes accumulation of the protein in axonal growth cones and results in aberrant axon morphology,29 which is consistent with a role of KIF21A in axonal growth cone regulation. As post-translational modification of tubulin is essential for regulation of primary cilia length and KIF21A inhibits microtubule growth at the cell cortex, disturbance of this pathway may be a cellular consequence of KIF21A deficiency and functional link to the phenotype of FA.
In summary, our clinical and genetic data from two FA families together with previous observations in S. scrofa provide firm evidence that while heterozygous gain-of-function variants in KIF21A cause CFEOM1, bi-allelic PTVs are associated with a severe form of FA in human. The observation of two independent events in a cohort of 23 index cases is compatible with pathogenic KIF21A variants representing a more common cause of FA. However, the rather small cohort size is a limitation of this study and genetic (re-)evaluation of larger FA cohorts is needed to estimate the contribution of this new disease locus to this collectively common disease entity and its relevance in the context of prenatal testing and the evolving diagnostic option of preconception carrier screening.
Data availability statement
Data are available upon reasonable request. All variants have been deposited into ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under Institute of Medical Genetics and Applied Genomics, University of Tübingen, including VCV… through VCV….
Ethics statements
Patient consent for publication
Ethics approval
The research project was approved by the Ethics Committee of the Medical Faculty of the University of Tübingen (number 066/2021BO2). All families provided consent according to the respective research protocols including patient photographs, approved by each of the institutional review boards (IRBs).
Acknowledgments
We thank the patients and their families contributing to this study. Furthermore, we thank Beate Kootz, Karin Hamann and Claudia Bauer for excellent technical support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
RJF and AJM contributed equally.
Contributors Conceptualisation—RF, AM and TBH. Data curation—RF, AM, MG and MS. Investigation—RF, AM, WK, MG, SS, PS, DG, AK, ND, EMCS, LA, UGr, RB, TH, NP, JP, MK, SH, DH, DE, NH, AN, GK, UGe, SHa, RS, SHe, MH, SO, SW, SB-W, DG, IT, OR, FD, KK, AD and TBH. Methodology—CS and OK. Software—MS and SO. Supervision—TBH. Visualisation—RF and MG. Writing (original draft)—RF, AM and TBH. Writing (review and editing)—RF, AM, MG and TBH. TBH is responsible for the overall content as guarantor.
Funding TBH was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation: 418081722, 433158657).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.