Article Text

Abstract
Background Developmental and epileptic encephalopathies (DEEs) represent a group of severe neurological disorders characterised by an onset of refractory seizures during infancy or early childhood accompanied by psychomotor developmental delay or regression. DEEs are genetically heterogeneous with, to date, more than 80 different genetic subtypes including DEE31 caused by heterozygous missense variants in DNM1.
Methods We performed a detailed clinical characterisation of two unrelated patients with DEE and used whole-exome sequencing to identify causative variants in these individuals. The identified variants were tested for cosegregation in the respective families.
Results We excluded pathogenic variants in known, DEE-associated genes. We identified homozygous nonsense variants, c.97C>T; p.(Gln33*) in family 1 and c.850C>T; p.(Gln284*) in family 2, in the DNM1 gene, indicating that biallelic, loss-of-function pathogenic variants in DNM1 cause DEE.
Conclusion Our finding that homozygous, loss-of-function variants in DNM1 cause DEE expands the spectrum of pathogenic variants in DNM1. All parents who were heterozygous carriers of the identified loss-of-function variants were healthy and did not show any clinical symptoms, indicating that the type of mutation in DNM1 determines the pattern of inheritance.
- epilepsy
- genetics
- nervous System Diseases
- Pediatrics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Developmental and epileptic encephalopathies (DEEs) represent a group of neurodevelopmental disorders characterised by epilepsy with an early childhood onset and severe psychomotor developmental delay or regression, and in some cases early death.1 DEEs are genetically heterogeneous with currently over 80 different subtypes enlisted in Online Mendelian Inheritance in Man (OMIM), defined by the underlying genetic defect.2 3 Most cases of DEE occur as sporadic events caused by autosomal dominant de novo variants, but autosomal recessive as well as X-linked inheritance patterns have also been observed.4
DNM1 encodes dynamin-1, a member of the dynamin-like family proteins. Dynamin-1 is a mechanochemical GTPase involved in clathrin-mediated endocytosis and endocytic vesicle fission.5 It is exclusively expressed in neurons and localises to the presynaptic terminal taking part in synaptic vesicle endocytosis and membrane recycling after neurotransmitter release.6 During endocytosis, dynamin molecules assemble into tetramers and form helical polymers at the necks of budding endocytosis vesicles.7 8 On GTP hydrolysis, these dynamin-1 helices undergo conformational changes that induce membrane constriction and subsequently result in vesicle fission from the membrane.9 10
In 2014, heterozygous missense variants in DNM1 (OMIM 602377) were first identified in five patients with early-onset DEE (DEE31, OMIM 616246).11 Since then, over 30 individuals with heterozygous, mostly de novo DNM1 variants were reported.11–18 All except for one reported variant, a de novo in-frame 6 bp insertion, are missense variants affecting highly conserved residues and predicted to exert a dominant-negative effect on dynamin-1 function.19 The mutational spectrum of DNM1 includes several recurrent variants. In approximately one-third of patients, a single recurrent variant in DNM1, p.Arg237Trp, has been identified.15 18 This variant affects a highly conserved arginine residue within the GTPase domain of DNM1 that is involved in conformational changes on GTP binding and, thereby, induces DNM1 multimerisation.
Here, we report two unrelated individuals with a severe neurological disorder characterised by early-onset epilepsy, generalised muscular hypotonia, visual impairment and severe neurodevelopmental delay. Using whole-exome sequencing (WES), we identified biallelic truncating variants in DNM1 in both individuals providing evidence that biallelic loss of DNM1 function is involved in the pathogenesis of DEE in humans.
Methods
Patients
Family 1 was identified at the Children’s Hospital, University Medical Centre Göttingen, Germany, and peripheral blood samples were collected from the index patient (III.1 in figure 1A) and both parents. Family 2 (figure 1B) was identified and clinically examined at the Centre of Clinical Genetics, Hadassah Hebrew University Hospital, Jerusalem, Israel. Peripheral blood samples of the affected individual, five clinically unaffected siblings and both parents were collected. DNA was extracted by standard extraction procedures. The Genematcher tool was used to connect the two centres in which the clinical and genetic analyses were performed.20

Pedigrees, clinical and genetic characteristics of individuals carrying biallelic variants in DNM1 pedigrees of family 1 (A) and family 2 (B) with pathogenic variants in DNM1. Affected siblings (solid symbols) in both families carry homozygous, truncating variants in DNM1, while non-affected parents are heterozygous carriers of the identified DNM1 variants (white symbols). Non-affected siblings were either homozygous for the WT allele or heterozygous carriers of the identified DNM1 variant. (C) Electropherograms of the identified DNM1 (ENST00000372923.8, NM_004408.3) variants c.97C>T (p.Gln33*) (upper panel) and c.850C>T (p.Gln284*) (lower panel). PCR and subsequent Sanger sequencing confirmed homozygosity of the identified DNM1 variants in both affected individuals and parental heterozygous carrier status. (D) Schematic representation of the DNM1 protein structure with an N-terminal GTPase domain (orange), a middle domain (blue), a pleckstrin homology (PH) domain (green), a GTPase effector domain (grey) and a C-terminal PRD (yellow). Black arrows indicate the localisation of the two truncating variants identified within this study. Localisation of heterozygous missense variants in DNM1 associated in previous studies with DEE are indicated by black asterisks.11–19 DEE, developmental and epileptic encephalopathy; GED, PH, PRD Pro-rich domain; WT, wild type.
WES and variant screening
In family 1, WES was performed on genomic DNA of the affected individual III.1. Exonic and adjacent intronic sequences were enriched using the Agilent SureSelectXT Human All Exon V7 enrichment Kit and were run on an Illumina HiSeq4000 sequencer at the Cologne Centre for Genomics (CCG), University of Cologne, Germany, and WES data analysis and filtering of mapped target sequences were performed with the ‘Varbank 2’ exome analysis pipeline (CCG, University of Cologne, Germany).
In family 2, we performed trio-based WES on DNA of the affected individual and both parents using the Agilent SureSelect Human All Exon 50 Mb V5 kit. Paired-end sequencing was carried out on an Illumina HiSeq2500 sequencer. Read alignment and variant calling were performed with DNAnexus (Palo Alto, California, USA) using default parameters with the human genome assembly hg19 (GRCh37) as reference.
All identified variants in DNM1 were confirmed by PCR amplification and subsequent Sanger sequencing on independent DNA samples and tested for cosegregation within the respective families.
Results
Clinical report
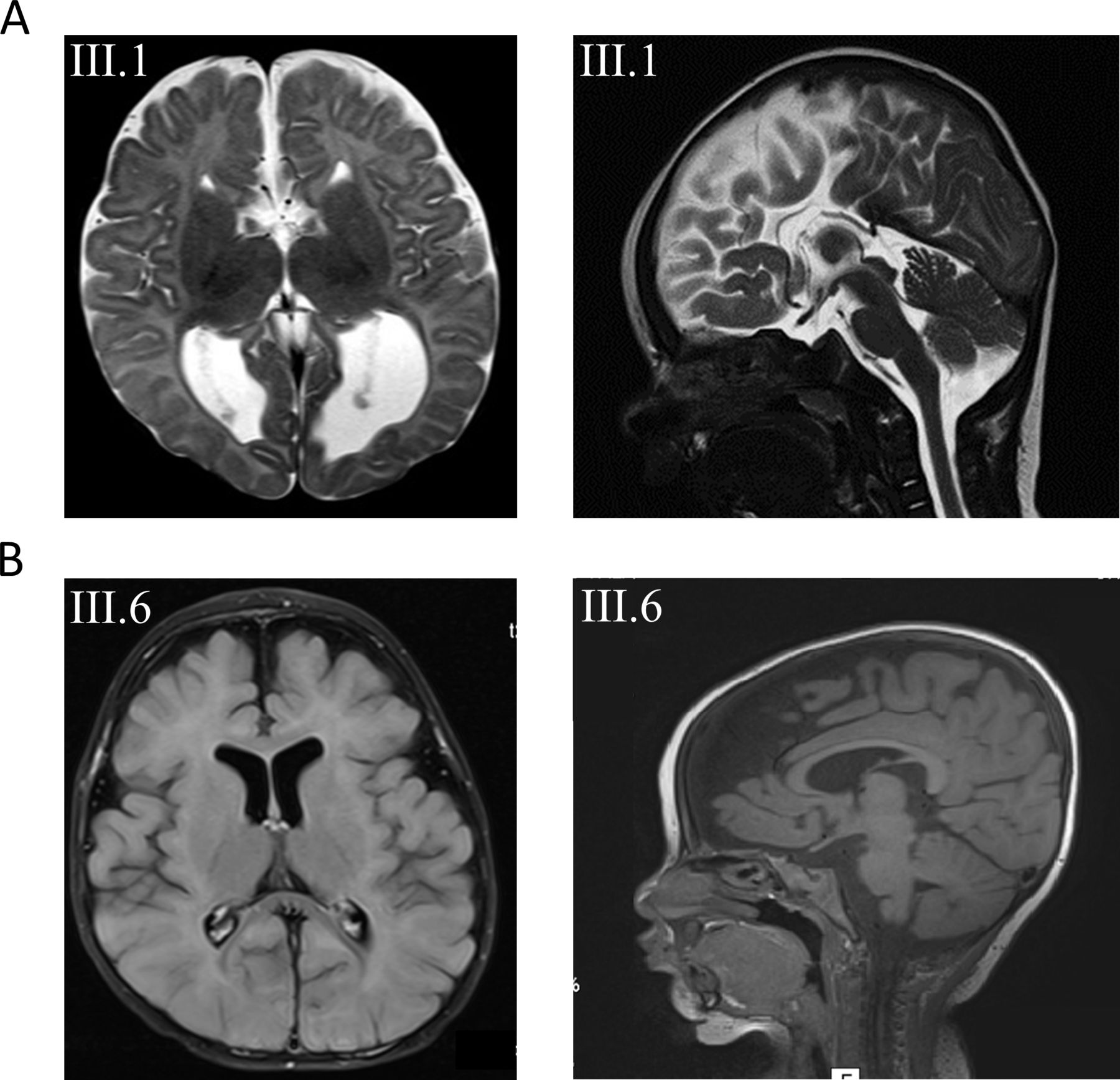
Family 1 is a consanguineous family of Lebanese origin (figure 1A). Individual III.1 is the first child of first-degree cousins. Both parents were healthy and did not show seizures or muscular hypotonia, and there is no history of developmental disorders or epilepsy in the family. Individual III.1 was born after an uneventful pregnancy at term with normal Apgar scores. Birth weight was 3540 g (+0.2 SD); length was 52 cm (+0.1 SD); and occipitofrontal circumference (OFC) was 35 cm (+0.1 SD). Routine transcranial ultrasound revealed agenesis of the corpus callosum (ACC) with colpocephaly. Apart from mild feeding problems, the first weeks of life were uneventful. At the age of 15 weeks, she presented with multifocal clonic seizures. Physical examination showed mild deceleration of head growth (OFC −0.7 SD), recurrent episodes of opisthotonus, impaired fixation and poor visual pursuit. Recurrent multifocal seizures were observed. Laboratory investigations of blood, urine and cerebrospinal fluid (CSF) provided no evidence for a metabolic disorder. Ophthalmological examination showed poor fixation, reaction only to bright light, but was otherwise normal, including normal funduscopy. EEG revealed multifocal epileptic discharges. Visual evoked potentials using flashing light from light-emitting diode goggles were not elicitable. MRI of the brain confirmed the ACC with colpocephaly but was otherwise normal (figure 2A). Pharmacotherapy with levetiracetam was started. She developed severe global developmental delay and secondary microcephaly during the first year of life. Generalised muscular hypotonia was mixed with dystonic movements of arms and legs. Frequent multifocal myoclonic and clonic seizures were treated with valproic acid, vigabatrin, topiramate and lamotrigine, with incomplete seizure control. At the age of 6 months, EEG showed hypsarrhythmia. Infantile spasms were not observed. Subsequent EEGs revealed multifocal spike-wave discharges and background slowing. At last follow-up at age 5 years, her body weight was 14.35 kg (−2.1 SD); length was 99 cm (−2.4 SD); and OFC was 45.8 cm (−4.4 SD). There were no dysmorphic features. In supine position, she showed sparse stereotypical spontaneous movements of arms more than legs, no turning, no sitting and hardly any head control. She showed some reaction to bright light but otherwise no fixation or visual pursuit. Hearing was normal. She reacted to familiar voices, but there was neither verbal understanding nor expressive speech.

{kind=link}
{kind=link}
(A) Axial and sagittal T2-weighted images of subject III.1 from family 1 at the age of 3 months revealing agenesis of the corpus callosum. (B) Axial and sagittal T1 images from subject III.6 from family 2 with mainly frontal brain atrophy, dilated subarachnoid spaced, widened perisylvian fissure and mildly dilated lateral ventricles.
Family 2 is a consanguineous family of Muslim–Arab origin (figure 1B). Individual III.6 was born after an uneventful pregnancy at 40 weeks of gestation by uncomplicated spontaneous delivery as the youngest of six children of first-degree cousins. Her siblings as well as her parents are healthy and did not present with seizures, muscular hypotonia or developmental delay. Her birth weight was 3080 g (−0.9 SD); birth length was 50 cm (−0.8 SD); and OFC was 34 cm (−0.7 SD). At the age of 4 months, sharp limb movements and irritability were noticed. At 6 months of age, she was hospitalised due to an increase in involuntary movements. She presented with infantile spasms, and EEG showed hypsarrhythmia. Treatment with vigabatrin and adrenocorticotrophic hormone (ACTH) was initiated. Subsequent therapy included prednisolone, topiramate, clonazepam, amlodipine and potassium supplementation. Brain MRI at 7 months of age showed mildly dilated ventricles and subarachnoid spaces, widened perisylvian fissure and frontal brain atrophy (figure 2B). Clinical examination at 1 year showed severe global developmental delay. She had a broad face, and we observed gingival hypertrophy. Additional dysmorphic features were not noted. Liver and spleen were normal. Axial and limb muscle tone were markedly reduced, but tendon reflexes were elicited. She was sleepy and showed decreased spontaneous limb movements. She was not able to roll over and hold objects, and did not make eye contact. Speech and language development was not observed. Auditory Brainstem Response (ABR) testing at 14 months of age was normal. At 2 years, she presented with microcephaly, visual disturbance and generalised muscular hypotonia. She had involuntary myoclonic jerks without EEG correlates. Due to failure to thrive and impaired swallowing, gastrostomy tube feeding was introduced at 2 years and 10 months of age. Eye examination at 3 years and 6 months revealed no pupillary light reflex or visual fixation, and we observed mild bilateral optic atrophy. She was not able to sit or lift her head. She had frequent myotonic limb movements with stereotypical movements of the upper limbs, and anticonvulsant treatment was continued with levetiracetam. At last follow-up at age 3 years 8 months, her weight was 10 kg (−3.7 SD); length was 88 cm (−3.1 SD); and OFC was 45 cm (−4.5 SD).
Mutation analysis
Chromosomal and cytogenomic microarray analyses did not reveal any likely pathogenic genomic alterations causative of the observed phenotype in both patients. To identify the underlying genetic cause of DEE, we performed WES of affected individuals from families 1 and 2. Parental samples were either analysed in parallel with the patient (family 2) or subsequently by Sanger sequencing (family 1). WES data were filtered for de novo, homozygous or compound heterozygous variants with an allele frequency ≥25%, and an MAF <0.5% in the gnomAD database,21 and we excluded any known pathogenic or likely pathogenic variants in all OMIM-annotated genes known for DEE. Based on parental consanguinity, we then prioritised homozygous exonic and/or splicing variants for further analysis, and we identified homozygous variants in only a single overlapping gene in both families predicted to have a severe impact on protein function and to be most likely deleterious (online supplemental tables 1 and 2). We identified two homozygous nonsense variants in dynamin-1 (DNM1; ENST00000372923.8, NM_004408.3), c.97C>T (p.(Gln33*)) in family 1 and c.850C>T (p.(Gln284*)) in family 2, which both led to an early stop and premature protein truncation (figure 1C,D). These DNM1 variants were heterozygous in each parent, and cosegregation analysis in family 2 showed wild-type or heterozygous carrier status for unaffected siblings (figure 1B). Both variants were absent from the gnomAD database, which in total contains only six loss-of-function variants, all in heterozygous state, annotated for DNM1, while 47 such variants were calculated to be expected in the >2 40 000 alleles (pLI=1).
Supplemental material
Discussion
In this study, we provide strong evidence that biallelic loss-of-function variants in the DNM1 gene cause a severe neurodevelopmental phenotype comprising early-onset epilepsy, muscular hypotonia, visual impairment and marked developmental delay. Since the first description of heterozygous missense variants in DNM1 as cause of epileptic encephalopathies in 2014,11 more than 20 exclusively missense variants and a single 2-amino-acid in-frame insertion were identified in patients with DEE, all except for one clustering either in the GTPase domain or the central middle domain of DNM1 (figure 1D).12–17 Functional analyses of the structural and cellular consequences of these missense variants most likely suggest a dominant negative effect on DNM1 function caused by impaired GTP hydrolysis/binding capability or directly affecting higher-order oligomerization of DNM1.15 22
Clinically, patients with heterozygous missense variants in DNM1 show relative phenotypical homogeneity with early-onset epilepsy, profound intellectual disability and hypotonia.15 Neuroimaging in these patients is usually normal or merely shows cerebral volume loss over time. Delayed myelination, thin corpus callosum, hypomyelination and Blake’s pouch cysts were observed in single cases.15 23 24 In line with these clinical characteristics, both of our patients present with severe global developmental delay, multifocal epilepsy and global hypotonia. Additionally, we observed severe visual impairment in both patients, which was also reported in previously published patients. Both developed secondary microcephaly. Neuroimaging revealed ACC in patient 1 and enlargement of inner and outer CSF spaces in patient 2 (figure 2). So far, structural brain anomalies including ACC have not been seen in patients with heterozygous DNM1 mutations. Online supplemental table 3 displays genetic, clinical and neuroimaging features of previously published patients with DNM1 encephalopathy associated with heterozygous mutations versus our two patients with biallelic mutations reported here.
Of note, all parents as well as siblings in family 2 carrying the identified loss-of-function variants in a heterozygous state are healthy. In line with this, heterozygous dynamin-1 knockout mice are viable and do not show any apparent anomalies, providing further evidence that haploinsufficiency is not the underlying pathomechanism of DNM1-associated DEE in humans.6 Similar to our patients, homozygous dynamin-1 knockout mice appeared normal at birth and were indistinguishable from their littermates. However, these animals developed poor motor coordination, showed reduced ingestion of milk and died within the first 2 weeks of life due to failure to thrive.6
In conclusion, we show that biallelic loss-of-function variants in DNM1 cause a severe neurodevelopmental phenotype characterised by early-onset epilepsy, generalised hypotonia, visual impairment and severe neurodevelopmental delay. The phenotype of patients with biallelic loss-of-function variants appears to be similar to those with heterozygous missense variants in DNM1, providing a further example of a gene in which the type of mutation and its functional effect determine the inheritance pattern and phenotypical outcome of the disease.
Ethics statements
Patient consent for publication
Ethics approval
This study was performed according to the Declaration of Helsinki protocols, and reviewed and approved by the local institutional ethics boards (University Medical Centre Göttingen, Germany; Hadassah Hebrew University Hospital, Jerusalem, Israel). Written informed consent was obtained from all affected subjects, parents or legal representatives participating in this study.
Acknowledgments
We are grateful to all patients and their families for their sustained cooperation, Christian Müller for excellent technical assistance and Karin Boss for critically reading the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
BW and KB are joint senior authors.
GY, RS and MD are joint first authors.
GY, RS and MD contributed equally.
BW and KB contributed equally.
Contributors GY, RS, BW and KB designed the study, and wrote and edited the manuscript, which was then approved by all coauthors; MD, RS and KB evaluated and examined families 1 and 2; GY, HMS, LD, YL, EK, JA, PN, SK and PB performed the experiments and analysed the data.
Funding This work was supported by the Israel Ministry of Health Exome Pilot Study, by the German Research Foundation (Deutsche Forschungsgemeinschaft) under Germany’s Excellence Strategy (EXC 2067/1-390729940) and the Collaborative Research Centres Programme SFB1002 (project D02 to BW), and by the Niedersächsisches Ministerium für Wissenschaft und Kultur (grant 74ZN1284 to KB).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.