Article Text

Abstract
Background Arthrogryposis multiplex congenita (AMC) is characterised by congenital joint contractures in two or more body areas. AMC exhibits wide phenotypic and genetic heterogeneity. Our goals were to improve the genetic diagnosis rates of AMC, to evaluate the added value of whole exome sequencing (WES) compared with targeted exome sequencing (TES) and to identify new genes in 315 unrelated undiagnosed AMC families.
Methods Several genomic approaches were used including genetic mapping of disease loci in multiplex or consanguineous families, TES then WES. Sanger sequencing was performed to identify or validate variants.
Results We achieved disease gene identification in 52.7% of AMC index patients including nine recently identified genes (CNTNAP1, MAGEL2, ADGRG6, ADCY6, GLDN, LGI4, LMOD3, UNC50 and SCN1A). Moreover, we identified pathogenic variants in ASXL3 and STAC3 expanding the phenotypes associated with these genes. The most frequent cause of AMC was a primary involvement of skeletal muscle (40%) followed by brain (22%). The most frequent mode of inheritance is autosomal recessive (66.3% of patients). In sporadic patients born to non-consanguineous parents (n=60), de novo dominant autosomal or X linked variants were observed in 30 of them (50%).
Conclusion New genes recently identified in AMC represent 21% of causing genes in our cohort. A high proportion of de novo variants were observed indicating that this mechanism plays a prominent part in this developmental disease. Our data showed the added value of WES when compared with TES due to the larger clinical spectrum of some disease genes than initially described and the identification of novel genes.
- genomics
- human genetics
- nervous system malformations
- neuromuscular diseases
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. All data relevant to the study are included.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Arthrogryposis multiplex congenita (AMC) is a developmental condition characterised by joint contractures in two or more body areas resulting from reduced or absent fetal movements. AMC has an overall incidence of 1 in 3000 to 5000.1 AMC is the direct consequence of reduced fetal movements which may lead, in addition to AMC, to pterygia, pulmonary hypoplasia, diaphragmatic defect or cleft palate.
There are multiple causes of AMC including (i) genetic defects, (ii) congenital infections with cytomegalovirus, varicella zoster virus, rubella virus and more recently Zika virus, (iii) extrinsic causes leading to limitations of fetal movements such as multiple pregnancy, oligohydramnios, amniotic bands or anatomical abnormalities of the uterus and (iv) maternal immune diseases such as myasthenia gravis.2
The genetic causes of AMC identified to date include a large spectrum of diseases which arise as a result of variants in genes encoding components required for the formation or the function of neuromuscular junctions, skeletal muscle, motor neurons, myelin of peripheral nerve, connective tissue of tendons and joints or central nervous system including brain with or without spinal cord anomalies.2 A total of 402 genes have been reported so far in AMC.3
There are multiple benefits for having a diagnosis and genetic evaluation for patients with AMC and their families. Indeed a specific diagnosis can precise the recurrence risks for relatives and is very helpful for the health surveillance and management required including physical therapy, orthopaedic treatment, and for the long-term prognosis.
Many AMC individuals remain without a genetic diagnosis suggesting the involvement of other pathogenic mechanisms or missed diagnosis owing to genetic heterogeneity. Our main goals were to improve the diagnosis rates of AMC, to evaluate the added value of whole exome sequencing (WES) compared with targeted exome sequencing (TES) and to identify new AMC genes. Here, we further explored genetic alterations in a cohort of 315 genetically undiagnosed and unrelated AMC families.
Patients and methods
Patients
A total of 315 unrelated families were included from 2011 to 2019 (online supplemental table S1). Inclusion criteria consisted of joint contractures in two or more body areas identified during pregnancy or at birth, without an unequivocal etiological diagnosis after clinical assessment by paediatricians, neuropaediatricians, fetal pathologists or clinical geneticists using targeted gene Sanger sequencing, chromosomal microarray (CMA) or molecular analysis of SMN1 (MIM: 600354) or DMPK (MIM:605377). The search for rubella or CMV infection was systematically performed. The parents of all affected individuals provided written informed consents for pathological examinations and genetic analyses of their affected children or fetuses and themselves in accordance with the ethical standards of our institutional review boards.
Supplemental material
Methods
Genetic mapping of disease loci
At the beginning of this study, genetic mapping of disease loci was performed in 15 multiplex and/or consanguineous families using Affymetrix GeneChip Human Mapping 250K NspI microarray. Multipoint linkage analysis of SNP data was performed using the Alohomora4 and Merlin software.5 In these families, genetic mapping was followed by either Sanger sequencing when a highly candidate gene was located within one of the disease loci (n=7) or by WES (n=8, figure 1).

Genomic approaches including genetic mapping, TES or WES. n: number of unrelated patients and in brackets the percentage of index patients with disease gene identification (Gene Id.). Other approaches (n=4) included CMA, SMN1, MTM1 or NIPBL analyses. TES, targeted exome sequencing; WES, whole exome sequencing.
Targeted exome sequencing (TES)
TES was performed on the DNA sample of affected individuals with AMC of unknown origin (n=210, figure 1). TES was performed using the Agilent SureSelectXT Custom kit (targeting 500 Kb including AMC genes and candidate genes for library preparation and exome enrichment). Sequencing was performed on an Illumina MiSeq System using paired-end 150 bp reads and following Illumina’s protocol using the MiSeq Reagent Micro Kit, V.2. The median coverage was 90×. Variants were selected using the same criteria as those used for WES data.
Whole exome sequencing (WES)
WES was performed from DNA of the index patient (n=209). WES used the Exome Capture Agilent SureSelect XT V5 kit for library preparation and exome enrichment as previously described6 in 123 patients. Sequencing was performed on a Genome Analyzer IIx Illumina instrument in paired-end mode with a read length of 2×100 bp. More recently, WES was performed using a completed Twist Bioscience Human Core Exome (Consensus CDS) kit for library preparation and exome enrichment in 86 patients (Integragen). Sequencing was performed on a Genome Analyzer Hiseq4000 Illumina instrument in paired-end mode with a read length of 2×80 bp (Integragen). The median coverage was 80×. WES was performed either after genetic mapping of multiplex and/or consanguineous families (n=8), or when the TES was negative (n=111) or directly (n=90, figure 1).
Bioinformatics analysis
Reads were aligned to the human reference genome sequence (UCSC hg19, NCBI build 37.3) via the BWA programme.7 Variants were selected using the SAMtools8 and then annotated using Annovar softwares.9 Variants in coding regions (including non-synonymous and nonsense variants), intron-exon junctions (≤10 bp) or short coding insertions or deletions were selected when the minor allele frequency was less or equal to 0.005 (using 1000G, ExAC, TopMed and GnomAD). Prediction of pathogenicity of missense variants was performed using Polyphen-2 (with score ≥0.510) or Sift softwares (with score ≤0.0511), splice variants using Human splicing finder12 and Clinvar (NCBI).
Sanger sequencing
Direct Sanger sequencing of candidate gene(s) located within the disease loci as established by genetic mapping was performed in seven families. Variants identified through either TES or WES were validated by Sanger sequencing. PCR amplification was carried out as previously described.6 PCR products were purified and then sequenced using the forward or reverse primers (Eurofins Genomics). The obtained DNA sequences were compared with published sequences (BLAST, NCBI). Sanger sequencing was also performed to establish the genotype of each family member and to analyse the segregation of variants within each family.
Other investigations
For recently identified genes, morphological analyses of skeletal muscle, neuromuscular junction or peripheral nerve were performed from patient samples and reported in separate reports.13–18 Functional validation of recently identified disease genes was investigated through the generation and characterisation of animal (C. elegans or Zebrafish) or cellular models and described in separate reports.13–18 When splicing mutations were identified, RNA analysis was performed to validate the pathogenic effects of mutations on RNA stability or exon splicing. Statistical analyses were performed using Fisher’s exact test two tailed.
Results
CLINICAL DATA
A total of 315 AMC families were included in this study from 2011 to 2019 (figures 1 and 2 and online supplemental table S1). AMC was sporadic in 226 families and familial (with at least two affected individuals) in 89 families (figure 2). In patients with sporadic AMC (n=226), 45 of them were born to consanguineous parents, 151 to non-consanguineous parents and unknown in 30 families. In familial AMC (n=89), parents were consanguineous in 30 families, non-consanguineous in 54 and unknown in 5 (figure 2).

Comparative analysis of the percentage of patients with unrelated AMC in whom the disease gene was identified depending on whether the AMC was (i) familial (at least two affected patients) or sporadic (section ‘All’), (ii) in sporadic patients born to either CSG parents or not, (iii) familial AMC born to CSG parents or not. Statistical analysis was performed using Fisher’s exact test two tailed. In sporadic or familial patients, the consanguinity was unknown in 30 or 5 families, respectively. AMC, arthrogryposis multiplex congenita; CSG, consanguineous; ns, not significant.
AMC was detected during pregnancy in 251 out of 315 families (80%), after birth in 62 families (20%) and not reported in 2 families (online supplemental table S1). During pregnancy, AMC was detected through ultrasound examination of the first trimester in 86 families (34%), second trimester in 103 families (41%) and third trimester in 27 families (11%). In 35 fetuses, the exact age of discovery of AMC during pregnancy was not reported.
The AMC was classified as non-syndromic versus syndromic depending on the identification of additional features not related to the fetal akinesia sequence. A total of 213 families was classified as non-syndromic (67.6%) and 102 as syndromic AMC (32.4%, table 1 and online supplemental table S1). The associated features were umbilical artery anomalies (n=3), brain involvement (including cognitive impairment, epilepsy, corpus callosum agenesis, perisylvian polymicrogyria, cerebellum hypoplasia, microcephaly, ventriculomegaly or macrocephaly, n=45), cardiac anomalies (such as cardiomyopathy or congenital heart defects, n=17), kidney anomalies (including unilateral or bilateral pyelectasia, unilateral renal agenesis, renal hypoplasia or urolithiasis, n=10), intrauterine growth retardation (n=30), bone agenesis (n=2), ears (such as hearing loss or unilateral ear hypoplasia, n=4) or eye anomalies (microphthalmia or cataract, n=5). Facial haemangioma was found in 12 affected individuals.
Summary of the main clinical features found in our cohort of 315 patients
Among syndromic AMC (n=102), both AMC and additional clinical features were discovered during pregnancy in 58 affected individuals (56.8%). In 19 affected individuals (18.6%), AMC and associated features were discovered after birth. Importantly, in 25 affected individuals (24.5%), AMC was diagnosed during pregnancy but not the associated features including brain anomalies in 17 of them (online supplemental table S1).
Genetic results
Genomic strategies and results
At the start of this study, genetic mapping of disease loci was performed in 15 families with at least two affected individuals or in sporadic affected individuals born to consanguineous parents. When a highly candidate AMC gene was identified within a disease locus, direct Sanger sequencing of the gene was performed. Otherwise, WES was performed in the index patient. The disease gene was identified in all patients (figure 1, online supplemental table S1) including seven recently identified disease genes (ADCY6 (MIM: 600 294), CNTNAP1 (MIM: 602 346), GLDN (MIM: 608 603), LGI4 (MIM: 608 303), LMOD3 (MIM: 616 112), MAGEL2 (MIM: 605 283) and UNC50 (MIM: 617 826), figures 1 and 3, (online supplemental table S1).13–18
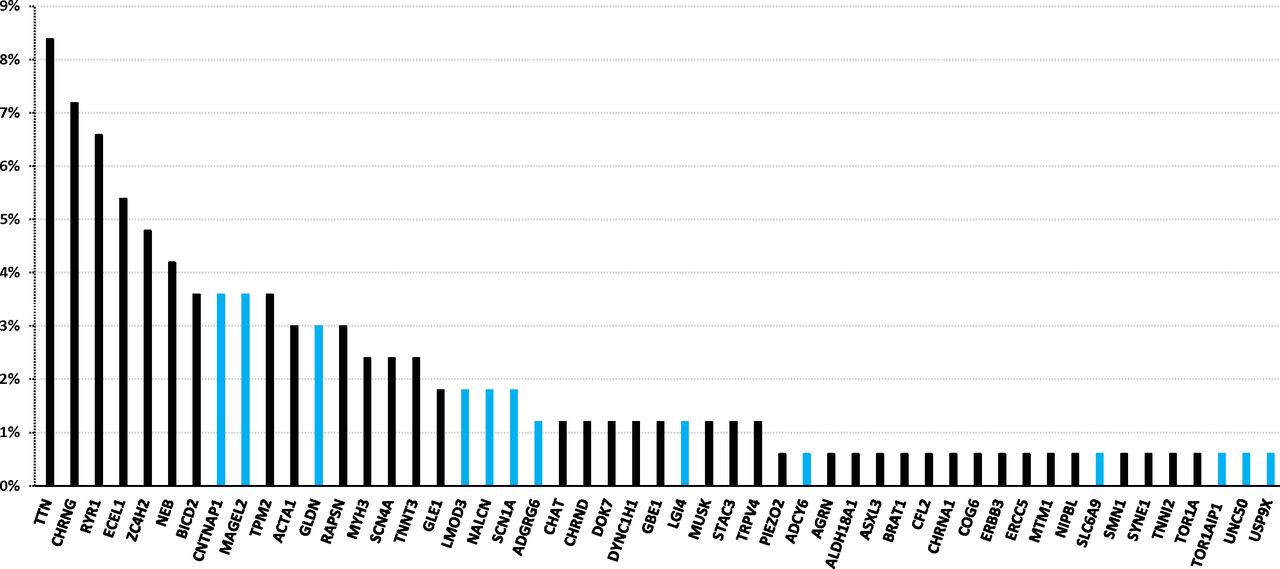
Genes in which pathogenic variants were identified in our cohort of AMC index patients. The percentage indicates the ratio of patients with unrelated AMC carrying pathogenic variant(s) in a given gene to 166, the total number of index patients with an identified disease gene. The blue colour indicates new genes identified in AMC within the last 6 years. AMC, arthrogryposis multiplex congenita.
Then, targeted exome sequencing of genes known to be frequently involved in AMC or new candidate genes was performed in 210 index patients (figure 1). The first panel included 67 genes and then moved to 84. The genetic cause was identified in only 68 affected individuals (32%) suggesting that other genes responsible for syndromic or non-syndromic AMC were not included in the panels, or novel genes not yet identified as responsible for AMC. In the patients with undiagnosed AMC, the phenotype was first re-evaluated based on the clinical data or muscle biopsy and led to other investigations including CMA, SMN1, MTM1 (MIM: 300415) or NIPBL (MIM: 608667) analysis which allowed the identification of the disease causing gene defect in four patients (see online supplemental table S1). In 27 negative TES, other investigations including WES were not performed because of the limited DNA quantity of the index patient. When no predicted pathogenic variants were identified through TES, WES sequencing was performed in 111 index patients (figure 1). WES allowed the identification of the disease causing gene in 24 index patients (21.6%). Among the genes identified, some of them were not included in the first TES gene panel such as BICD2 (MIM: 60979719), DYNC1H1 (MIM: 60011220), GBE1 (MIM: 60783921), SCN4A (MIM: 60396722) and NALCN (MIM: 61154923) which have been shown to be responsible for AMC since 2013. Others are genes responsible for disease for which joint contractures were not reported as the main clinical features such as ASXL3 (MIM: 61511524), STAC3 (MIM: 61552125), USP9X (MIM: 30007226) and three recently identified genes including GLDN (MIM: 60860314), LGI4 (MIM: 60830315) and SCN1A (MIM: 182389,27 figure 3). In seven index patients, WES allowed the identification of pathogenic variants in ACTA1 (MIM: 102610), AGRN (MIM: 103320), MUSK (MIM: 601296), TPM2 (MIM: 190990), TTN (MIM: 188840) and ZC4H2 (in two patients, MIM: 300897) due to bad coverage of the variants through the TES panels (online supplemental table S1).
WES was therefore performed in the first line in 90 index patients and the disease gene was identified in 55 of them (61.1%, figure 1). This strategy allowed us to confirm four recently identified AMC causing genes including CNTNAP1, MAGEL2, ADGRG6, SCN1A 13 17 27 28 or genes recently identified by other teams including TOR1AIP1 (MIM: 61451229) and SLC6A9 (MIM: 601019,30 online supplemental table S1 and figure 3).
Altogether, the disease gene was identified in 166 out of 315 index patients (52.7%) and the number of disease genes found in our AMC cohort was 51 (figure 3). New genes recently identified in AMC in the last 6 years were found in 35 index patients and represent 21.1% of disease causing genes in our cohort (CNTNAP1, MAGEL2, GLDN, LMOD3, SCN1A, ADGRG6, LGI4, UNC50, ADCY6, SLC6A9, NALCN and TOR1AIP1, figure 3). In addition, we showed that pathogenic variants in ASXL3 and STAC3 might be responsible for early onset motor defect leading to AMC as the first clinical symptoms. Variants in ASXL3 were reported in Bainbridge-Ropers syndrome which is characterised by delayed psychomotor development and severe intellectual disability (MIM: 61548524). Variants in STAC3 are responsible for Bailey-Bloch congenital myopathy (MIM: 25599525) and were more recently reported in AMC.31 Variants in USP9X are known to be responsible for female-restricted X linked syndromic mental retardation (MIM: 30096826). Our data indicate a critical role of these genes in prenatal motor development leading to AMC broadening the phenotypic spectrum of variants in these genes.
Comparing the efficiency of TES with that of WES, the most efficient approach is WES (61.1% of disease causing genes identified in affected individuals) when compared with all TES performed (32%, figure 1).
Pathogenic mechanisms in syndromic and non-syndromic AMC
This study allowed an evaluation of the pathogenic mechanism through the function of the identified gene (figure 4). The most frequent cause was a primary involvement of skeletal muscle in 40.6% of index patients (n=67), brain with or without spinal cord involvement in 22.4% (n=37), neuromuscular junction in 17% (n=28), axoglial interaction in 10.3% (n=17) and spinal cord in 9.1% (n=15). The disease genes known to be associated with brain involvement including or not spinal cord and found in our cohort were ALDH18A1 (MIM: 138250), ASXL3, BICD2, BRAT1 (MIM: 614506), CNTNAP1, COG6 (MIM: 606977), ERCC5 (MIM: 133530), MAGEL2, NALCN, PIEZO2 (MIM: 613629), SCN1A, SLC6A9, TOR1A (MIM: 605204), USP9X and ZC4H2 (online supplemental table S1).

Pathogenic mechanisms in AMC. This study allowed an evaluation of the pathogenic mechanism through the function of the identified gene. The percentage indicates the ratio of patients carrying a mutation in a group of genes involved in a given function to 165, the total number of index patients with an identified disease gene (without the patient carrying the chromosomal translocation). AMC, arthrogryposis multiplex congenita; NMJ, neuromuscular junction.
The disease causing gene was identified in 40 out of 102 patients with syndromic AMC (39.2%) while in non-syndromic AMC (n=213), the disease gene was identified in 126 patients (59.1%, Fisher’s exact test, two-tailed p=0.0011, (online supplemental table S1). In syndromic AMC, there was no significant difference in gene identification between consanguineous (42.3%, 11/26) and non-consanguineous AMC (41.5%, 27/65, p=1). There was also no statistically significant difference in disease gene identification in patients with syndromic AMC when the phenotypic characterisation was based on prenatal data only (15 out of 47 patients, 31.9%) when compared with the one based on postnatal findings (25 out of 55 patients, 45.5%, p=0.22).
Interestingly, after delivery, a total of 12 patients with AMC (12 out of 146 index patients, 8.2%) displayed facial angioma. Among them, variants in ECEL1 (MIM: 605896), CHRNG (n=2, MIM: 100730), BICD2 and RAPSN (MIM: 601592) were found in 5 patients (online supplemental table S1). In 7 out of 12 patients with AMC with facial haemangioma, genomic analysis did not identify the gene defect. In four of them, in addition to facial haemangioma, AMC was associated with other clinical features such as mental retardation, macrosomia, cholestasis or congenital heart defect (online supplemental table S1). This data indicate that AMC associated with facial haemangioma is clinically and genetically heterogeneous including the subgroup of amyoplasia.
Comparative analysis of genomic results in familial versus sporadic, consanguineous versus non-consanguineous AMC patients
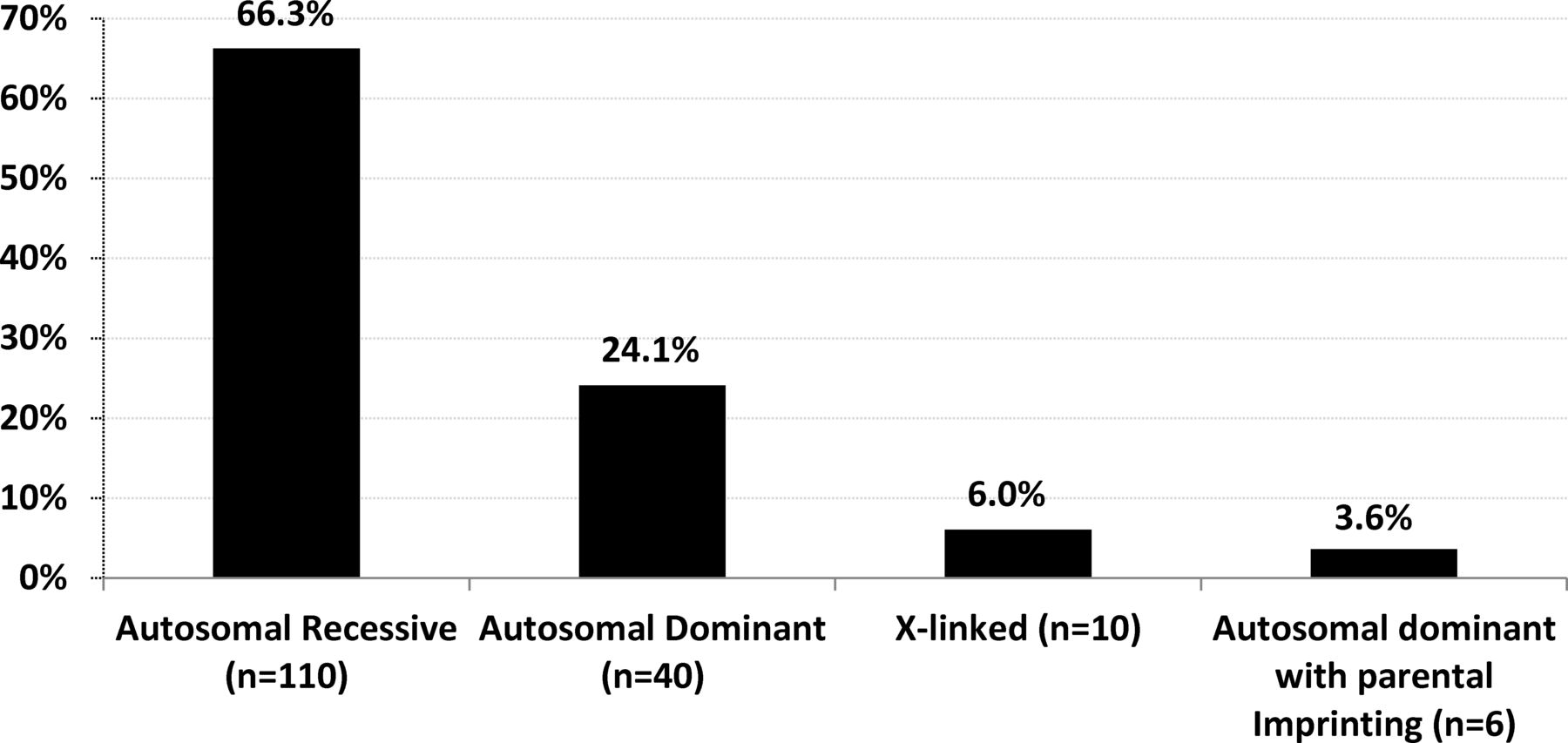
The identification of disease causing genes in 166 AMC families allowed a better evaluation of the distribution of modes of inheritance (figure 5). In 40 index patients (24.1%), the mode of inheritance was autosomal dominant with 70% of de novo variants. It was autosomal recessive in 110 index patients (66.3%), X linked in 10 (6%) with a de novo variant in 6 of them and autosomal dominant with parental imprinting in 6 index patients (3.6%) with MAGEL2 mutations with a de novo variant in 4 of them. Altogether, the percentage of de novo variants was 22.9% (38 out of 166). The most frequent mode of inheritance of AMC is autosomal recessive which represents 66.3% of diagnosed patients in our cohort.

Modes of inheritance based on disease gene identification. The percentage indicates the ratio of patients carrying pathogenic variants (s) with a given mode of inheritance (n) to 166, the total number of index patients with an identified disease gene.
We then compared the number of AMC families in whom the gene was either identified or not depending on whether the AMC was familial or sporadic and in sporadic patients born to consanguineous parents or not (figures 2 and 6). There was a highly statistically significant difference in gene identification in familial AMC (66 out of 89, 74%) when compared with sporadic AMC (100 out of 226, 44%, Fisher’s test, p<0.0001). In sporadic AMC, there was also a statistically significant difference in gene identification in patients born to consanguineous parents (29 out of 45, 64%) when compared with those born to non-consanguineous parents (60 out of 151, 40%, Fisher’s test, p=0.0039, figure 2).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
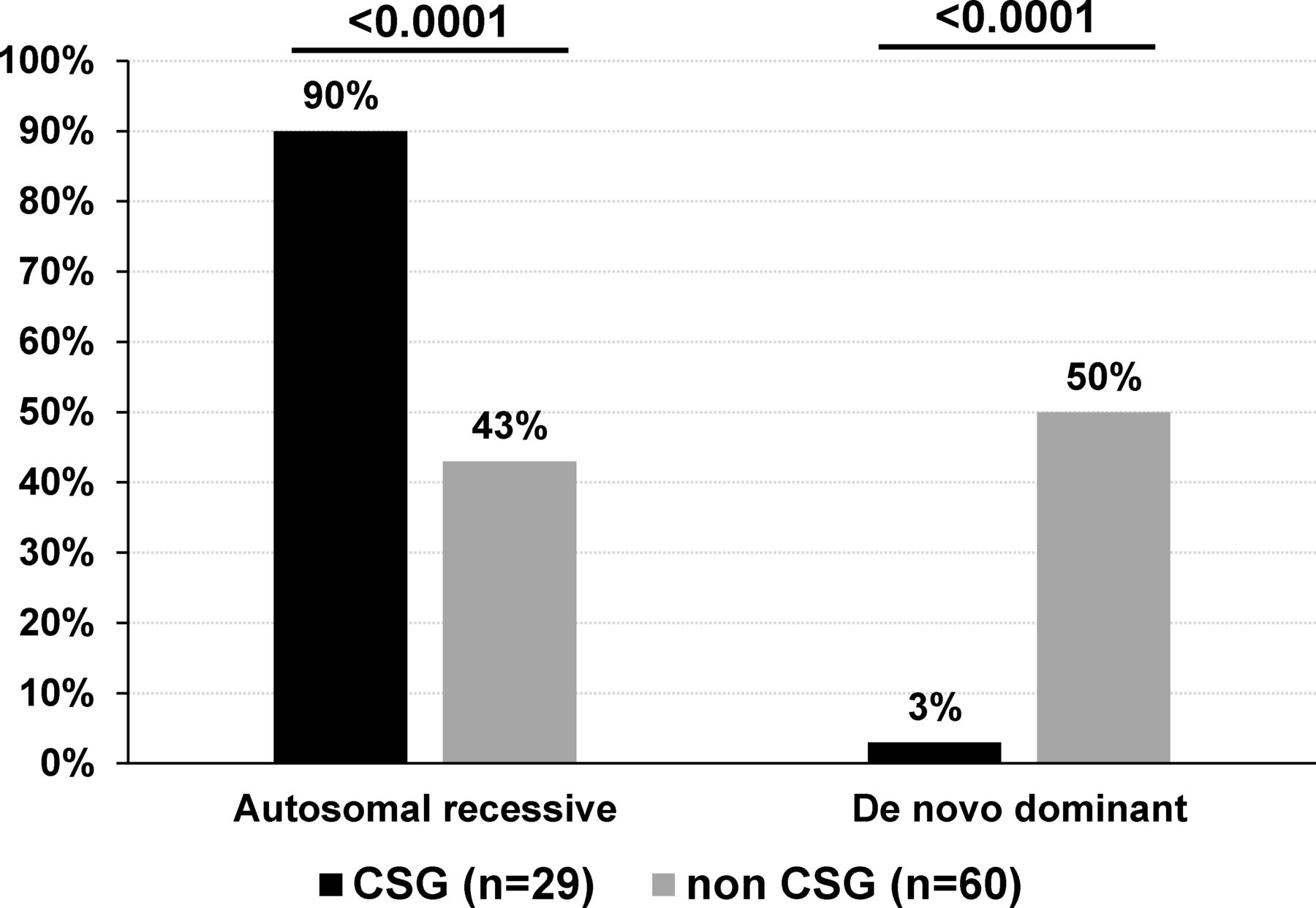
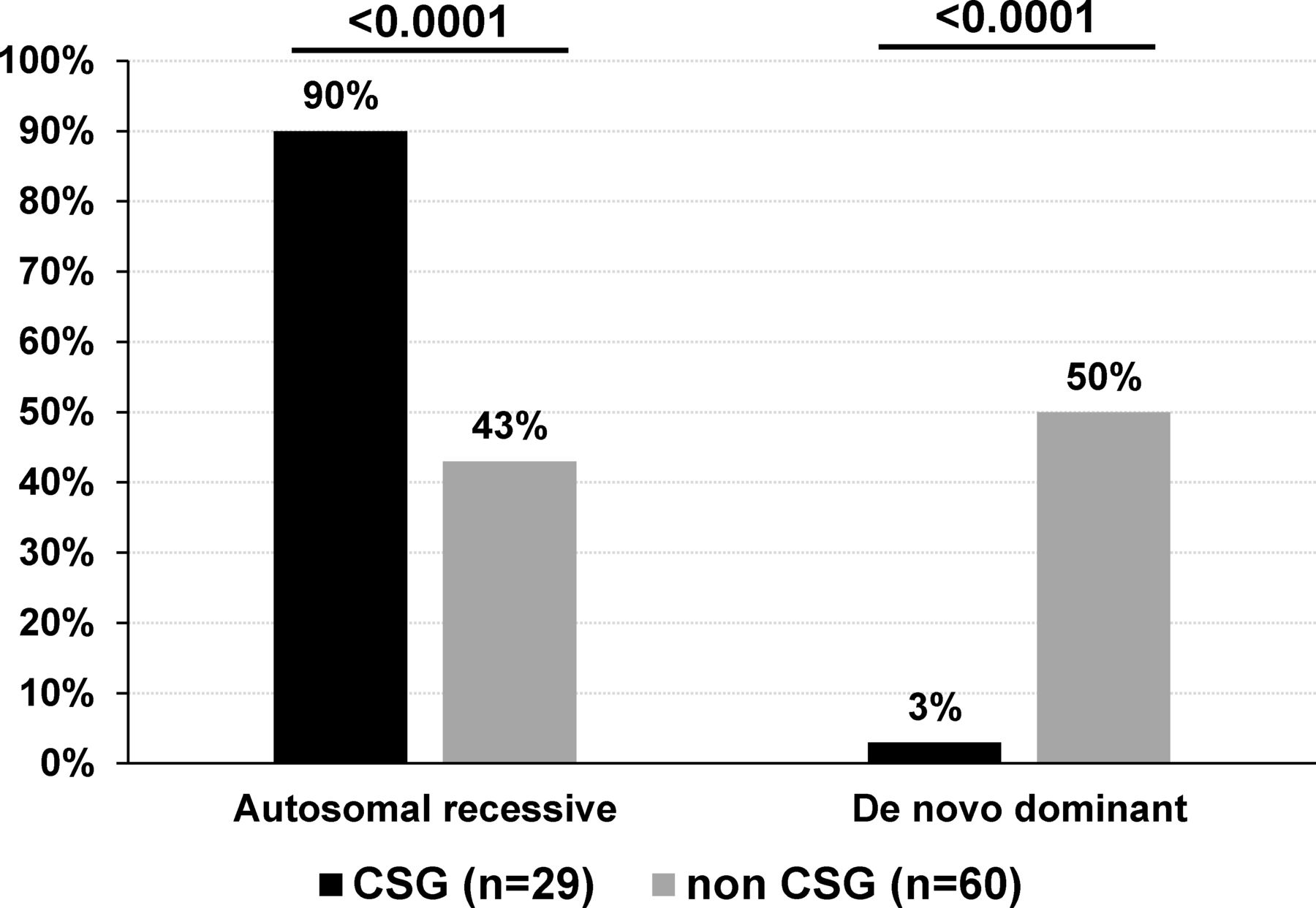
Modes of inheritance of sporadic patients with AMC based on disease gene identification. De novo mutations include AD, AD with parental imprinting or X linked modes of inheritance. The percentage indicates the ratio of AMC with autosomal recessive or de novo mutation to the total number of CSG (n=29) or non-CSG index patients (n=60) with an identified disease gene. Statistical analysis was performed using Fisher’s exact test two tailed. AD, autosomal dominant; AMC, arthrogryposis multiplex congenita; CSG, consanguineous.
Among sporadic patients and based on gene identification, the most frequent mode of inheritance was autosomal recessive in patients born to consanguineous parents (89.6%, 26 out of 29 patients with homozygous variants in 25 of them) when compared with 43.3% (26 out of 60) in patients born to non-consanguineous parents (Fisher’s test, p<0.0001, figure 6). Importantly, de novo dominant autosomal or X linked variants were observed in 30 out of 60 sporadic patients born to non-consanguineous parents (50%) when compared with 3% in sporadic patients born to consanguineous parents (1 out of 29 patients, Fisher’s test, p<0.0001, figure 6). Although in 15 multiplex and/or consanguineous families, genetic mapping allowed the identification of disease loci, WES or direct Sanger sequencing detected pathogenic variants similar to those found using TES or WES only indicating that these highly statistically significant differences in gene identification in multiplex or consanguineous AMC families compared with non-consanguineous patients with sporadic AMC are not caused by the nature or the position of the identified variant.
Discussion
Pipis et al reported that in Charcot Marie Tooth disease (CMT), WES is a valuable research tool, with independent groups reporting diagnostic rates of 19%–45% in individuals with CMT or complex neuropathy for whom previous genetic testing was negative.32 Similarly, Ghaoui et al reported that using WES, pathogenic variants were identified in 45% of patients with limb-girdle muscular dystrophy.33 In AMC, a conclusive genetic diagnosis was established in 47% in an Australian cohort of 38 families34 suffering from fetal akinesia/hypokinesia, arthrogryposis or severe congenital myopathies and 58.3% including candidate genes in a Turkish cohort of 48 AMC families.35 More recently, Ravenscroft et al performed next generation sequencing in 190 probands and 81 of them received a genetic diagnosis (42.6%).31 In our cohort of 315 AMC families, the largest one reported to date, our results indicate that WES is the most efficient approach with an ability of disease gene identification in 61% of AMC index patients. Our data showed indeed the added value of WES when compared with TES due to the larger clinical spectrum of some disease genes than initially described and the identification of recently published novel genes.
Indeed, this strategy allowed the identification of new AMC disease mechanisms which have been published as separate reports such as proteins involved in node of Ranvier formation (CNTNAP1, GLDN),13 14 axoglial interaction for peripheral myelination (ADCY6, ADGRG6, LGI4),13 15 28 AChR trafficking (UNC50),18 organisation of sarcomeric thin filaments in skeletal muscle (LMOD3),16 brain development (MAGEL2)17 and Na+ channel function in motor cortex (SCN1A).27 Other recently identified disease genes such as TOR1AIP1,29 SCL6A9 30 or NALCN 23 were also found in our AMC cohort. Altogether, genes recently identified since 2014 in either non-syndromic or syndromic AMC were found in 21.1% of disease causing genes in our cohort. In addition, we identified pathogenic variants in ASXL3,24 STAC3,25 USP9X 26 genes in patients with AMC broadening the phenotypic spectrum of variants in these genes. Variants in TTN, CHRNG, RYR1 and ECEL1 genes were found in 46 out of 166 AMC index patients in whom the disease gene was identified (27.7%) representing therefore the most prevalent genetic causes of AMC (figure 3). Our results confirmed the marked genetic heterogeneity in AMC. Indeed, a total of 51 disease genes were identified in our cohort and for 21 of them, pathogenic variants were identified in a single family. New candidate genes have been recently reported in autosomal-recessive AMC35 36 but no predicted or possibly pathogenic variants in these candidates were identified in our cohort.
In 47.3% of AMC index patients, a genetic cause was not established. Whole genome sequencing (WGS) has shown its superior diagnostic and analytical sensitivity to WES owing to its ability to assess SNVs (single nucleotide variation), indels and CNVs (copy number variation) in coding and non-coding regions and more complete per-base coverage. WGS yield has not been evaluated in AMC to date and should be proposed when the genetic cause is not established. WES or TES analysis of the index patient and not trio is likely a weakness of our approach for the detection of de novo variants in new genes. Nevertheless, the reanalysis of WES in our cohort of undiagnosed patients allowed the selection of predicted pathogenic variants in shared genes than the analysis of the intrafamilial segregation through Sanger analysis. This allowed the recent identification of de novo heterozygous variants of SCN1A in three unrelated AMC index patients.27 One hypothesis for unidentified disease causing gene would be the involvement of a digenic mechanism. However, when looking at possibly pathogenic heterozygous variant in a given gene known to be responsible for AMC, we did not observe a recurrent association with a variant in another known AMC gene. An alternative hypothesis could be the occurrence of somatic mosaicism either dominant or recessive in combination with germinal allelic variant. Further investigation including the genetic analysis of affected tissues from patients could help answer this hypothesis. Another hypothesis to explain also the marked difference between either consanguineous or familial AMC and sporadic AMC is that in numerous patients, the cause of AMC is not genetic such as possibly novel maternal immune disease(s) or viral infection such as recently described in AMC caused by Zika virus infection which might be added to rubella and CMV screening in AMC.37
Importantly, the analysis of this large cohort revealed that, based on the identified gene, the most frequent cause of AMC was a primary involvement of skeletal muscle in 40.6% of index patients (n=67) followed by brain involvement with or without spinal cord defect which represents 22.4% of AMC (n=37). Among syndromic AMC (n=102), in 25 affected individuals, ultrasound examinations during pregnancy were normal except AMC. Among them, brain involvement was identified after birth in 17 index patients and variants in BICD2, CNTNAP1, MAGEL2, NALCN, SCN1A, SLC6A9 or ZC4H2 genes were found in 9 of them. Therefore, during pregnancy and when the AMC is observed through ultrasound examinations, the identification of the disease gene using a large gene panel or WES is critical for parent information especially when brain involvement is predicted or not through gene identification. Altogether, central nervous system involvement was observed in 44 out of 315 AMC individuals (14%) and was characterised by either isolated or associated neurologic symptoms including, for the most frequent symptoms, epilepsy (n=9), intellectual disability (n=16) or brain malformations (n=19, online supplemental table S1).
We found that the contributions of de novo and recessive variants were quite different among consanguineous and non-consanguineous sporadic patients with AMC based on gene identification. As expected, an autosomal-recessive inheritance is the most frequent mode of inheritance in sporadic patients born to related parents (90%) when compared with 43% in sporadic patients born to unrelated parents. Importantly, de novo coding variants were observed in 50% of sporadic patients born to unrelated parents (when compared with 3% in patients born to related parents) and recessive coding variants in 43%. Interestingly, the contribution of de novo dominant variants was quite similar in sporadic patients with AMC born to unrelated parents (50%) to that reported in probands with European ancestry from the large Deciphering Developmental Disorders Study (DDD, 49.9%,38). In the DDD study, 88% had an abnormality of the nervous system including intellectual disability or autism. Therefore, even the phenotypes of their patients were quite different from the one reported here, a similar and high proportion (~50%) of de novo variants was observed indicating that de novo dominant mutations play a prominent part in pathogenic mechanism of developmental diseases.
The benefits of an accurate genetic diagnosis include a better understanding of prognosis, more tailored management of AMC and possibly other organ involvement and improved surveillance. A precise genetic diagnosis of AMC enables an accurate genetic advice to the affected individuals and their families and may provide them with increased reproductive choice, for example, by enabling preimplantation diagnosis, non-invasive prenatal testing or prenatal diagnosis.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. All data relevant to the study are included.
Ethics statements
Patient consent for publication
Acknowledgments
We thank all families for partaking in this study. We thank the French Society of Foetal Pathology (SoFFoet) for referring clinical data and tissues of fetal patients. We thank D. Gaillard (CHU de Reims), F. Girard (CHU de Strasbourg), F. Giuliano (CHU de Nice), M. Lebrun (CHU de Saint Etienne), A. Masurel (CHU de Dijon), M. Mathieu (CHR Amiens), N. Monnier (CHU Grenoble), M. J. Perez (CHRU de Montpellier) and J. Roume (CH Poissy) for contribution and/or analysis of patient samples.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors AL designed the study with JMe, phenotyped the patients, performed morphological analysis of the neuromuscular system and contributed to writing the manuscript. DJ, EA, JMa, DM, AVi, KD, RS, LQ, FN, VB, PLat and DS conducted molecular analyses. IG conducted WES in 123 patients. KD, YC, AVe, BB, LLa, TA-B, JMa, SB, FPet, CB, SW, FM, DH, GV, JA, DA, CB, MB, LF, P-SJ, SK, SS, A-LD, AG, M-LJ, LLo, VL, SL, AM, LVM, JP, FG, PLan, PLet, FPel, LP, M-HS-F, HT, LT, CV-D, HA, CB, AB, EB, EB-B, VC-D, AD-D, ID, BE, CF, SG, DL, FL, ML, DM-C, AM, SM, MN, LR, FPr, CQ, HR, NR, AT, HV, MV, EC, CF-B, MG, RG, JS, MG, CG recruited and phenotyped the patients. J-B, AG-M, VB, PLat and DS validated the variants for diagnosis purpose. J-LB and MT generated and characterised animal models. JMe designed the study with AL, performed bioinformatics analysis of next generation sequencing data and wrote the manuscript. The authors contribute to the recruitment of patients or performed genomics analyses.
Funding This work was supported by a grant from the French Ministry of Health (PHRC 2010, AOM10181), the Association Française contre les Myopathies (AM, DAJ1891), the Agence de Biomédecine (2016), the Alliance Arthrogrypose and the Institut National de la Santé et de la Recherche Médicale (Inserm) to JMe. Several authors of the manuscript are members of the European Reference Network for Developmental Anomalies and Intellectual Disability (ERN-ITHACA).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.