Article Text

Abstract
Background Triploidy is one of the most common chromosome abnormalities affecting human gestation and accounts for an important fraction of first-trimester miscarriages. Triploidy has been demonstrated in a few cases of recurrent pregnancy loss (RPL) but its molecular mechanisms are unknown. This study aims to identify the genetic cause of RPL associated with fetus triploidy.
Methods We investigated genomic imprinting, genotyped sequence-tagged site (STS) markers and performed exome sequencing in a family including two sisters with RPL. Moreover, we evaluated oocyte maturation in vivo and in vitro and effect of the candidate protein variant in silico.
Results While features of hydatidiform mole were excluded, the presence of triploidy of maternal origin was demonstrated in the fetuses. Oocyte maturation was deficient and all the maternally inherited pericentromeric STS alleles were homozygous in the fetuses. A deleterious missense variant (p.V1251D) of the cyclin B3 gene (CCNB3) affecting a residue conserved in placental mammals and located in a region that can interact with the cyclin-dependent kinase 1 or cyclin-dependent kinase 2 cosegregated in homozygosity with RPL.
Conclusion Here, we report a family in which a damaging variant in cyclin B3 is associated with the failure of oocyte meiosis II and recurrent fetus triploidy, implicating a rationale for CCNB3 testing in RPL.
- copy-number
- reproductive medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Recurrent pregnancy loss (RPL) affects up to 5% of clinical pregnancies1 and is associated with both parental and embryonal factors including chromosomal abnormalities, endometrial changes, intrauterine abnormalities, endocrinological dysfunctions, infections and immunological abnormalities.2 3 Numerical chromosome abnormalities in the conceptus are the most common cause of early miscarriages,4 and triploidy accounts for about 13% of these cases.5 Triploid conceptuses can arise through three main mechanisms: (1) fertilisation of an oocyte by two spermatozoa; (2) fertilisation of an oocyte by a diploid spermatozoon; (3) fertilisation of a diploid oocyte.4 The last mechanism gives rise to a digynic triploid embryo and occurs more frequently as consequence of retention of the second polar body during meiosis, but can also be caused by retention of the first polar body or endoreduplication within the maternal pronucleus.6 A few cases of recurrent (3+) digynic triploidy have been reported, suggesting a genetic predisposition, but no conclusive results on identification of the causal gene(s) have been achieved so far.7–11 Recurrent dispermic triploidy has also been rarely described and associated with the histopathological characteristics of partial hydatidiform mole.12
RPL may also result from defective genomic imprinting.13 This is a mechanism causing the expression of a restricted number of genes to depend on their parental origin, due to differential epigenetic modification of the maternal and paternal alleles. Loss of function variants of proteins belonging to the subcortical maternal complex (SCMC) affect establishment of methylation of imprinted loci in oocytes and/or its maintenance in early embryos and are associated with infertility, RPL and abnormal embryo development.
Cyclins are regulatory subunits that by binding and activating their catalytic partner serine-threonine kinases (cyclin-dependent kinases, CDKs) control eukaryotic cell division and play pivotal roles in male and female meiosis.14 B3-cyclins contain motifs characteristics of A- and B-type cyclins, but are evolutionary distinct from the others.15 In placental mammals, cyclins B3 are larger (about 1400 residues) because of the presence of a long sequence derived from an additional exon between the N-terminal destruction box (residues 60–68) and the C-terminal cyclin boxes (residues 1132–1375).16 Although not completely defined, the function of cyclin B3 appears to be distinct from that of the other cyclins.17 In chicken somatic cells, cyclin B3 is able to bind to both CDK1 and CDK2.18 Human cyclin B3 has been shown to interact with CDK2 in vitro, but the complex displays low kinase activity.19 Mouse cyclin B3 is able to interact with CDK1 and its inactivation leads to female infertility associated with failure of maternal meiosis I and embryonic triploidy, but has no consequence on somatic cell division and male gametogenesis.20 21 However, misexpression of the cyclin B3 gene (Ccnb3) interferes with meiosis II arrest and exit during oogenesis through interaction with CDK2.22
In this study, we describe a family with RPL associated with digynic triploidy in the progeny. Genotyping of polymorphic sequence-tagged sites (STSs) and analysis of whole-exome sequencing (WES) indicate failure of maternal meiosis II associated with a homozygous damaging variant in the cyclin B3 gene (CCNB3 [MIM: 300 456]) that possibly caused the triploidy in the fetuses.
Materials and methods
A family (figure 1) including two sisters with RPL was referred to the Royan Reproductive Center (Tehran, Iran) in 2015. Peripheral blood samples were collected from the affected individuals II-6 and II-7, their partners II-5 and II-8, their parents I-3 and 1–4 and their siblings II-9 and II-10. Paraffin-embedded biopsies were collected from the products of conception III-12 and III-15 derived from miscarriage pregnancies of II-7. Detailed information about karyotyping, array comparative hybridisation, histological analysis of fetal tissue, oocyte retrieval and in vitro maturation (IVM), whole-exome sequencing, in silico protein modelling, as well as analyses of DNA methylation, STSs and variant segregation is given in online supplementary Materials and Methods. All participants provided informed consent after receiving information about the study procedure and aims.
Supplemental material

Pedigree of the family. Two women with RPL history (II-6 and II-7) were born from first-cousins (I-3 and I-4). The probands (II-6 and II-7) and their parents (I-3 and I-4) underwent WES after routine clinical procedure failing to determine the cause of their RPL. Analysis of WES data revealed that II-6 and II-7 were homozygous for the missense variant g.50346749T>A (ChrX, GRCh38/hg38) (SCV000886503, p.V1251D) in the CCNB3 gene. The genotypes of II-5, II-8, II-9, II-10, III-12 and III-15 were determined by Sanger sequencing. RPL, recurrent pregnancy loss; WES, whole-exome sequencing.
Results
Two sisters with RPL (II-6 and II-7, figure 1), who were born from consanguineous marriage between first-cousins (I-3 and I-4), were referred to our institute. II-6 had a history of 6 miscarriages and II-7 experienced 16 consecutive miscarriages. Clinical evaluation of uterine anatomy, and coagulation, hormonal, immunological and microbiological testings showed no sign of abnormality in either II-6 and II-7 (online supplementary table 1). All miscarriages occurred during the first 12 weeks of pregnancy in both affected sisters. The other family members did not report any history of RPL. Karyotype testing of the probands (II-6 and II-7) and their husbands (II-5 and II-8) showed normal chromosome patterns (46, XX and 46, XY) (online supplementary figure 1A-D). Also, no clinically significant chromosomal imbalance was detected by array CGH/SNParray in both couples (II-5 and II-6, and II-7 and II-8) and a product of conception (III-12) of II-7 (online supplementary figure 1E-H). However, it must be taken into account that arrayCGH and SNP array do not detect polyploidy, balanced translocations and inversions.23 Histological examination performed on III-12 demonstrated the presence of normal fetal chorionic villi along with central stroma covered by cytotrophoblast and syncytiotrophoblast, thus excluding any evidences of hydatidiform mole, trophoblastic proliferation, cellular atypia and central cistern formation (online supplementary figure 1J).
To investigate the status of genomic imprinting in the fetuses, the methylation level of the differentially methylated regions (DMRs) of seven imprinted loci that are located on different chromosomes and whose methylation abnormalities are associated with RPL and SCMC variants13 was determined in III-15 by bisulfite conversion and pyrosequencing. Compared with controls, we found that the PLAGL1, MEST, GRB10, KCNQ1OT1 and GNAS-AS1 DMRs that are normally methylated on the maternal chromosome were hypermethylated, and the H19/IGF2 and MEG3 DMRs that are normally methylated on the paternal chromosome were hypomethylated in III-15. These results excluded a defect of imprinting establishment or maintenance, but were consistent with the presence of genome-wide digynic triploidy in this fetus (online supplementary figure 2 and online supplementary table 2).
Triploidy of maternal origin was confirmed in fetuses III-12 and III-15 by STS marker genotyping, which also excluded endoreduplication within the maternal pronucleus as cause of the triploidy (online supplementary table 3). To determine if the digynic triploidy derived from an error in meiosis I or an error in meiosis II, we evaluated the segregation of pericentromeric STSs (<10 Mbp from centromere). We found homozygosity of maternal alleles for 10/10 informative markers in III-15 and for 3/3 informative markers in III-12, consistent with failure of meiosis II in maternal oocytes (table 1).
Segregation of pericentromeric STSs
Oocytes were retrieved from II-6 for in vitro fertilisation (IVF). Of 10 oocytes retrieved after one ovarian stimulation cycle, 7 were at MI stage and 3 at GV stage and no MII oocytes were observed, compared with an average of 85% MII oocytes usually obtained from a controlled ovarian stimulation cycle.24 IVM was also deficient; all oocytes failed to mature and were discarded during IVF procedures, compared with an average of 80% oocytes reaching the MII stage during normal IVM.25 To overcome the fertility problem, egg donation was proposed to II-6, as part of IVF. With egg donation, II-6 got pregnant and gave birth to two twin boys, further supporting the hypothesis that the cause of infertility was in oocyte development.
In order to determine the causal genetic variations underlying digynic triploidy, WES was performed on four individuals (I-3, I-4, II-6 and II-7) of the family. Considering the history of consanguinity, we postulated that the RPL phenotype was transmitted through a recessive pattern and the pathogenic variant was present in homozygosity in the affected individuals. After filtering and exclusion of frequent variants, two homozygous variants were found in both the probands (II-6 and II-7) (online supplementary table 4). The first of these variants consisted in an in-frame 3 bp deletion in a polymorphic CAG repeat (22 CAG, compared with a range of 11–36 and an average of 20–23 repeats in normal populations) of the androgen receptor gene.26 Thus, only the second variant affecting the X-linked cyclin-B3 (CCNB3) gene was retained as possible causal factor of RPL in this family. The homozygous missense variant g.50346749T>A (p.V1251D) (ChrX, GRCh38/hg38; SCV000886503) in exon 10 of CCNB3 was identified in II-6 and II-7 (figure 2). The variant that is present in the cyclin box—cyclin dependent kinase (CDK) binding domain of CCNB3 was not listed in gnomAD and dbSNP databases and was predicted to be deleterious by SIFT, probably damaging by PolyPhen and disease-causing by Mutation Taster prediction tools (table 2). Furthermore, V1251 is relatively conserved in placental mammals: the isosteric Val and Thr residues are present in primates and non-primates, respectively, in this position (online supplementary figure 3). Segregation of the candidate variant was investigated in 10 family members by bidirectional Sanger sequencing. The results showed that, in addition to II-6 and II-7, the variant was present in homozygosity in another sister (II-9) who had no pregnancy yet, in hemizygosity in their father (I-3) and in heterozygosity in their mother (I-4) and aborted fetuses (III-12 and III-15) (figure 1).

The g.50346749T>A (p.V1251D) (ChrX, GRCh38/hg38; SCV000886503) variant of the CCNB3 gene. (A) Schematic representation of the CCNB3 gene derived from UCSC (hg38) showing the sequences coding the destruction box (yellow) and the cyclin box domain (red) in NM_033031. Coding exons are indicated by dark blue boxes and intronic regions by blue lines. The transcription orientation is indicated with arrowheads. The g.50346749T>A is located in exon 10 (NM_033031) in the cyclin box region. (B) Example of Sanger sequencing confirming the segregation of the CCNB3 variant with the phenotype in II-6 and I-4. The variant (T4050A) is in red and showed on the forward strand.
Prediction of V1251 effects on CCNB3 function and structure
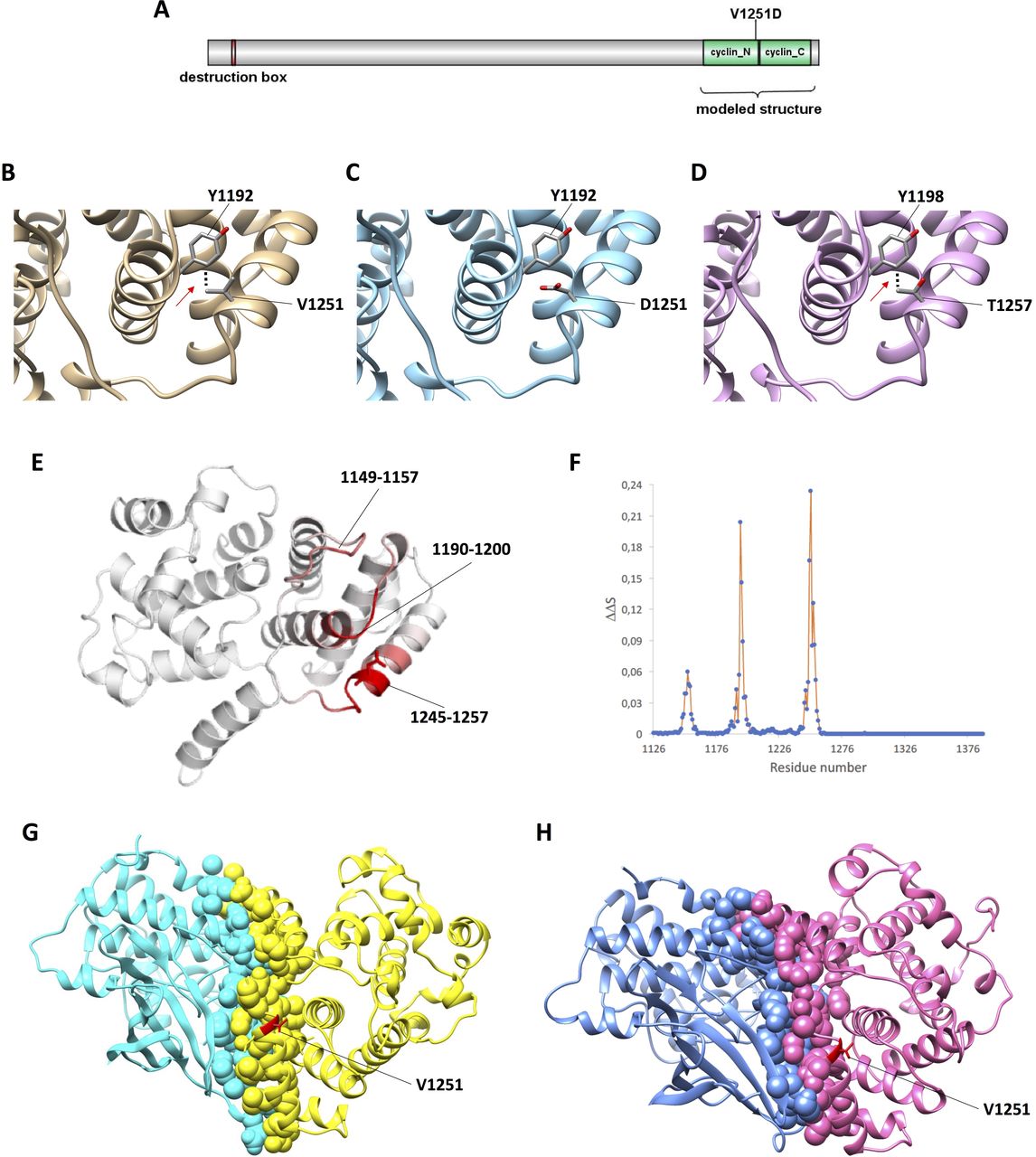
The structure of cyclin-B3 has not been determined yet. Approximately 1000 amino acid residues at the N-terminus are classified as disordered regions and only the destruction box (residues 60–68), and cyclin boxes (1132–1257 and 1259–1375) can be aligned to the other cyclins. Thus, the segment spanning from position 1126 to 1388 of human cyclin B3 was aligned to human cyclin-B1 (CCNB1) of which structures were available,27 28 and homology models of the wild-type as well as V1251D-mutant CCNB3 were built (figure 3A–C). We analysed the amino acids in close contact with the site of mutation (<4 Å) and observed that on the basis of this model, valine 1251 was located on the protein surface making interactions with tyrosine 1192 (figure 3B). V1251D results in the replacement of the hydrophobic side chain of valine with the negative side chain of aspartic acid. This leads to disruption of local intramolecular interactions with Tyr 1192 (figure 3C). Interestingly, this interaction is conserved in the mouse protein, in which T1257 replaces human V1251 and Y1198 replaces human Y1192 (figure 3D). In both cases, an interaction occurs between the methyl group on the beta carbon of valine or threonine and the aromatic ring of tyrosine. Moreover, to generate an optimised and robust predictor effect of V1251D substitution on protein stability and dynamics, we used DynaMut, a comprehensive suite evaluating the effect of variants by implementing a consensus estimate of changes on protein folding free energy calculated by different methods, such as ENCoM and DUET (including mCSM and SDM).29 Consistent with the changes of intramolecular interactions demonstrated in the model, the programme DynaMut predicted that the variant V1251D destabilises and increases the vibrational entropy (ie, molecular flexibility) of human CCNB3 (table 2). This result is graphically illustrated in figure 3E,F. According to DynaMut, the major changes in molecular flexibility (depicted in red in the model) in mutant CCNB3 occur in residues 1149–1157, 1190–1200, 1245–1257. Thus, the destabilising effect of V1251D is directly exerted on the interacting Tyr 1192 and indirectly on the region 1149–1157 (figure 3E,F).

{kind=link}
{kind=link}
{kind=link}
Effect of the V1251D variant on CCNB3 structure as predicted by in silico studies. (A) Linear representation of CCNB3. The domains destruction box (residues 60–68), and cyclin N (1132–1257) and cyclin C (1259–1375) boxes are depicted. (B–D) Effect of the V1251D variant on CCNB3 structure as predicted by in silico modelling. Close-up of the variant in human wild-type (B), human mutant (C) and mouse wildtype (D) CCNB3. The model of CCNB3 structure is shown as a cartoon, and the human Val/Asp 1251 and Tyr 1192, and mouse Thr 1257 and Tyr 1198 as sticks coloured by atom type. The hydrophobic bonds between Val/Thr and Tyr are indicated by dots. (E–F) Effect of the V1251D variant on CCNB3 structure as predicted by DynaMut. The changes of vibrational entropy (ΔΔS) of each residue in mutant vs wild-type CCNB3 are indicated as coloured protein model (E) and as chart (F). In the model, red represents a gain in flexibility. The figures were produced with the Dynamut webserver. (G–H) Complex between the CCNB3 cyclin box and CDK1 (G) or CDK2 (H) obtained by in silico docking showing Val 1251 close to the interface. CCNB3 is in yellow in (G) and pink in (H), CDK1 is in cyan in (G) and CDK2 is in blue in (H). Atoms at the interface are represented as spheres, Val 1251 is in red and its side chain is represented as sticks.
The partner kinase of human cyclin B3 in vivo is unknown.17 The modelled CCNB3 was docked onto the structures of human CDK127 or CDK2.28 We found that the solutions with the lowest energy were clustered and the relative orientations of the cyclin and kinases closely resembled those experimentally observed in the CCNB1-CDK1 and CCNB1-CDK2 complexes (online supplementary figure 4). The model reveals that V1251 is located at the end of a long alpha-helix (residues 1238–1251) and importantly is located just in between two regions (L1249-N1250 and K1253-I1258) interacting with the kinase (figure 3G,H).
Discussion
In this study, we report a novel homozygous missense mutation in the CCNB3 gene as a possible causative agent of RPL associated with fetal digynic triploidy. In silico studies predicted that the variant V1251D is deleterious for CCNB3 structure and function. The mutation disrupts an intramolecular hydrophobic interaction conserved in the mouse and destabilises the protein. In addition, the position of valine 1251 in the protein structure suggests that its substitution with aspartic acid affects CCNB3-CDK interaction. Finally, segregation of STS markers in the family indicates that triploidy resulted from failure of maternal meiosis II.
Cyclin B3 has been implicated in the control of female meiosis in different organisms. Females of D. melanogaster lacking cyclin B3 are infertile and their oocytes are unable to complete meiosis I, but no defect is evident in their ovary structure.30 In addition, cyclin B3 is downregulated in the ovaries of striped bass females producing poor quality eggs.31 In C. elegans embryos, cyclin B3 promotes anaphase onset in meiosis II and mitosis through interaction with the spindle assembly checkpoint and dynein.32 Mouse Ccnb3 inactivation causes female infertility and failure of oocyte metaphase-anaphase transition and extrusion of the first polar body in meiosis I.20 21 Consistent with our results, fertilisation of mouse Ccnb3 -/- oocytes by intracytoplasmic sperm injection produced triploid embryos that died before embryonic day 7.5 (equivalent to human first trimester).20 Studies in model organisms also indicated that CCNB3 is dispensable during male gametogenesis.20 21 Similarly, in our study, the male I-3 carrying the V1251 CCNB3 variant in hemizygosity is normally fertile. These sex-specific differences may be due to the longer duration of female meiosis I or compensation by other cyclins. Also, in both human and mouse, down-regulation of CCNB3 does not seem to have a negative impact on embryo development. In apparent contrast with our results, SNP genotyping of embryonic stem cells derived from parthenogenetic activation of mouse Ccnb3 -/- oocytes suggested that Ccnb3 inactivation affects segregation of homologous chromosomes in maternal meiosis I.20 It is possible that this discrepancy is due to differences between the knockout and the V1251D variant that destabilises CCNB3 but probably does not completely abolish its function or to differences between mouse and human meiosis. On the other hand, a recent study indicates a possible role of mouse cyclin B3 in completion and exit from oocyte meiosis II that is exerted through interaction with CDK2.22 Consistent with this report, our study indicates that human oocytes carrying the CCNB3-V1251D variant can correctly complete recombination and chromosome segregation in meiosis I but fail to segregate sister chromatids in meiosis II after in vivo fertilisation, resulting in embryonic triploidy.
We investigated the effect of the V1251D variant on cyclin B3 structure and function by modelling the cyclin B3 box. First, we observed that the methyl group on the beta carbon of valine 1251 interacts intramolecularly with the aromatic ring of a tyrosine and that this interaction is conserved in the mouse, where threonine, an isosteric amino acid with an analogous methyl group on the beta carbon, takes the place of valine. In the mutant protein, the substitution of valine with the negatively charged aspartic acid completely abolishes this interaction. In addition, the predicted changes in protein dynamics indicated that the mutant protein is less stable than the wild type; hence, it is expected that the intracellular concentration of V1251D CCNB3 is lower than normal. Finally, we evaluated CCNB3 interaction with either CDK1 or CDK2 by protein docking. It is worth considering that the structures of CDK1 and CDK2 are very similar.27 The computational results showed that Val1251 is on the protein surface and occupies a pivotal place immediately adjacent to two regions interacting with the kinases. We therefore predict that, by disturbing the concentration of cyclin B3 or its affinity for CDK1 or CDK2, the V1251D variant affects the correct completion of oocyte meiosis II and sister chromatids segregation, resulting in embryonic triploidy. Another example of how cyclin B3 can affect cell cycle regulation has been reported in human pathology and is provided by the Ewing-like sarcoma, in which an oncogene is formed by the fusion between the BCOR and CCNB3 genes.33
In summary, this study highlights CCNB3 as a maternal-effect gene possibly involved in the pathogenesis of RPL associated with fetus triploidy and suggests to include it in future investigations on this clinical condition.
Accession number
The accession number for the variant c.T4050A reported in this paper is [ClinVar]: [SCV000886503].
Acknowledgments
We thank the patients and their family members for their collaboration. We thank Mariano Rocchi for precious advice on STS analysis and David Asher for great help in interpretation of DynaMut results.
References
Footnotes
Contributors MT designed and conceptualised the study, NF, NS, LP, PP, MVC, FR, PR, MV, FR-S, AB, AS, BA, OP and MC performed analysis and interpreted the data. AR and MT drafted the manuscript. All authors critically revised the manuscript, contributed significantly to this work and declare to meet the ICMJE criteria for authorship.
Funding This work was supported in part by a grant from Royan Institute, Iran; Université Grenoble Alpes, France and Italian MIUR-PRIN 2015 JHLY35 (AR and MVC); Telethon-Italia GGP15131 (AR), Associazione Italiana Ricerca sul Cancro IG 2016 N.18671 (AR); 'Progetti per la ricerca oncologica della Regione Campania' Grant: I-Cure (AR) and 'Progetti competitivi intra-Ateneo' Programma VALERE (VAnviteLli pEr la RicErca) 2019 Grant: MIRIAM, Università degli studi della Campania 'Luigi Vanvitelli' (AR and AS). AS was supported by Programma VALERE (VAnviteLli pEr la RicErca) from Università degli studi della Campania 'Luigi Vanvitelli'.
Disclaimer The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study was approved by the Royan Institute ethics committee and conducted in accordance with the declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.