Article Text

Abstract
Background ZNF597, encoding a zinc-finger protein, is the human-specific maternally expressed imprinted gene located on 16p13.3. The parent-of-origin expression of ZNF597 is regulated by the ZNF597:TSS-DMR, of which only the paternal allele acquires methylation during postimplantation period. Overexpression of ZNF597 may contribute to some of the phenotypes associated with maternal uniparental disomy of chromosome 16 (UPD(16)mat), and some patients with UPD(16)mat presenting with Silver-Russell syndrome (SRS) phenotype have recently been reported.
Methods A 6-year-old boy presented with prenatal growth restriction, macrocephaly at birth, forehead protrusion in infancy and clinodactyly of the fifth finger. Methylation, expression, microsatellite marker, single nucleotide polymorphism array and trio whole-exome sequencing analyses were conducted.
Results Isolated hypomethylation of the ZNF597:TSS-DMR and subsequent loss of imprinting and overexpression of ZNF597 were confirmed in the patient. Epigenetic alterations, such as UPD including UPD(16)mat and other methylation defects, were excluded. Pathogenic sequence or copy number variants affecting his phenotypes were not identified, indicating that primary epimutation occurred postzygotically.
Conclusion We report the first case of isolated ZNF597 imprinting defect, showing phenotypic overlap with SRS despite not satisfying the clinical SRS criteria. A novel imprinting disorder entity involving the ZNF597 imprinted domain can be speculated.
- DNA methylation
- genetics, medical
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Genome imprinting is an epigenetic marking mechanism that causes genes to be expressed in a parental-origin-specific manner.1 Allelic expression patterns of clusters of imprinted transcripts are coordinately regulated by an imprinting control region (ICR). To date, all ICRs in humans overlap with germline differentially methylated regions (gDMRs), also known as primary DMRs, that exhibit parental allele-specific DNA methylation inherited from gametes and maintained throughout subsequent somatic development.2 gDMRs are essential in establishing additional somatic DMRs (sDMRs; also known as secondary DMRs) within the imprinted domains during development, leading to the additional allele-specific epigenetic features in somatic cells.2 3
Many studies have shown that imprinted genes exert fundamental effects on mammalian development and growth, and thus primary disruption of the methylation state at imprinted genes is associated with imprinting disorders in humans. In this context, Silver-Russell syndrome (SRS; OMIM 180860) is one of the representative imprinting disorders characterised by prenatal and postnatal growth retardation and some dysmorphic features. Primary molecular causes of SRS are loss of methylation on chromosome 11p15.5 (seen in 30%–60% of patients) and maternal uniparental disomy (UPD) of chromosome 7 (UPD(7)mat; 5%–10% of patients).4 Besides, pathogenic copy number variants (CNVs) and other imprinting disorders, such as Temple syndrome and maternal UPD of chromosome 20 (UPD(20)mat), are occasionally detected. In addition, it is notable that we and others have recently determined that maternal UPD of chromosome 16 (UPD(16)mat) is associated with the development of SRS.5 6
The most consistently identified and well-described imprinted locus on chromosome 16 is the human-specific, paternally imprinted (maternally expressed) zinc-finger gene ZNF597 located on 16p13.3. In humans, the paternally methylated sDMR named ZNF597:TSS-DMR was identified in the shared promoter region of the ZNF597 and NAA60 genes, which presumably regulates the maternal expression of both ZNF597 and NAA60 (figure 1A).7 8 The neighbouring ZNF597:3′ DMR is a gDMR functioning as an upstream regulator for the methylation pattern of the ZNF597:TSS-DMR.8–10 Whereas the biological functions of ZNF597 and NAA60 remain to be clarified, we have suggested that excessive expression of ZNF597 in patients with UPD(16)mat might contribute to their phenotypes, in particular growth failure.6
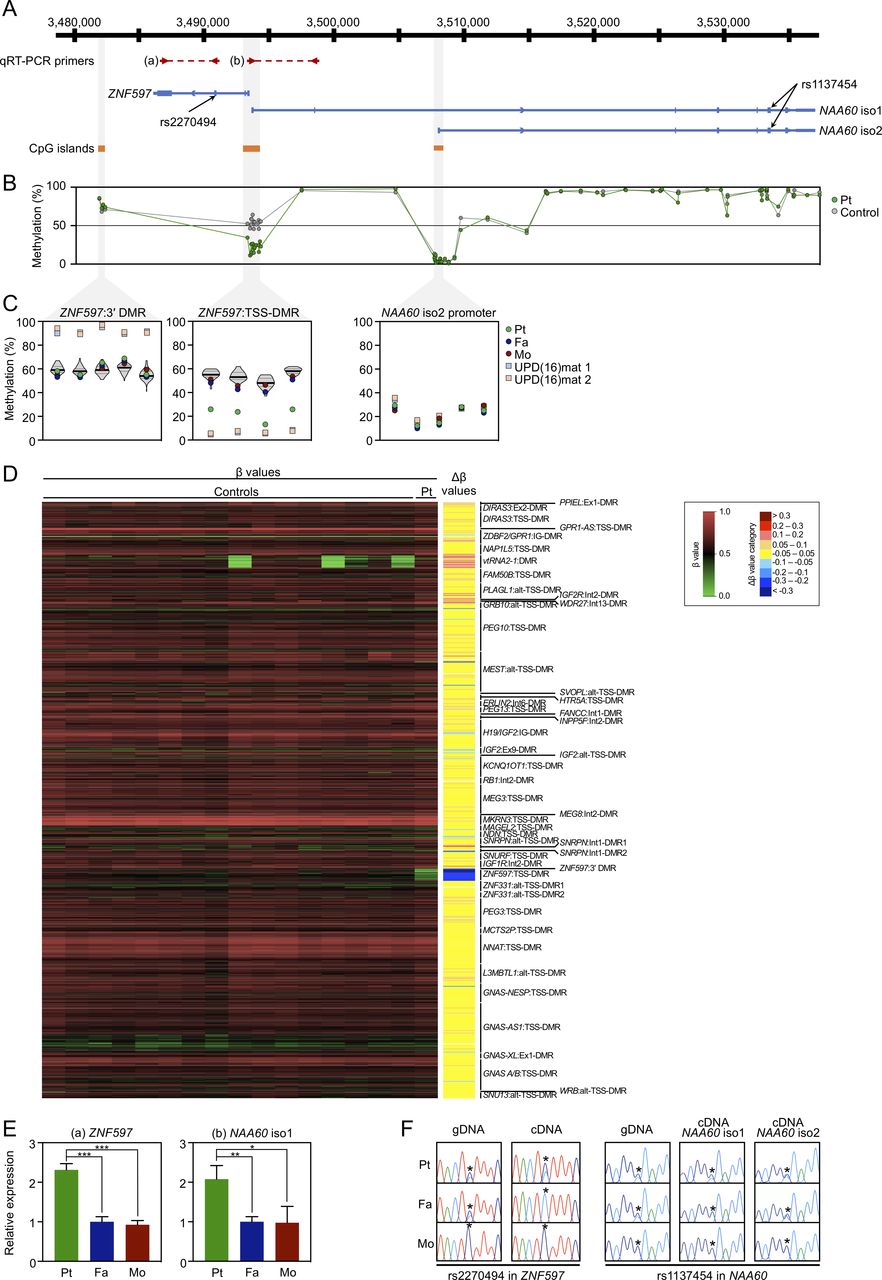
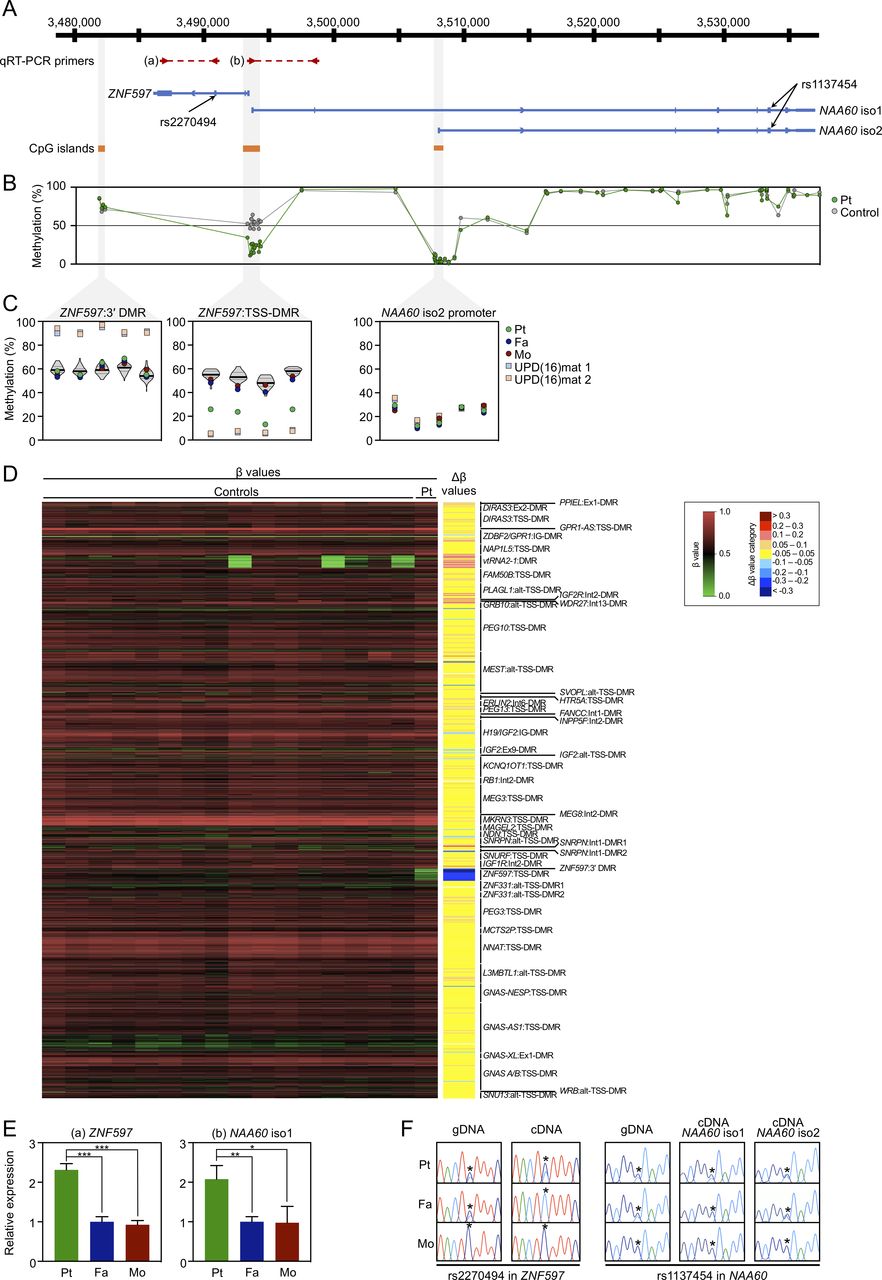
Methylation and expression analyses of the ZNF597/NAA60 imprinted domain in leucocyte samples. (A) Schematic representation of the domain, depicting the position of the qRT-PCR primers, transcripts, heterozygous SNPs and CpG islands with hg19 genomic coordinates. (B) Methylation levels of 73 CpG sites across the ZNF597/NAA60 imprinted domain by the Infinium MethylationEPIC BeadChip analysis for the leucocytes of the patient (green dots) and 16 control subjects (grey dots; average values are shown). (C) Methylation levels by pyrosequencing for the leucocyte genomic DNA from the patient (green dots), father (blue dots) and mother (red dots) as well as patient 1 (light blue squares) and patient 2 (light orange squares) with UPD(16)mat described in our previous report.6 Violin plots in the ZNF597:3′ DMR and ZNF597:TSS-DMR show the distribution of methylation levels of 55 control subjects, including the medians (bold lines) and IQRs (dotted lines). Five CpG sites in the ZNF597:3′ DMR, four sites in the ZNF597:TSS-DMR and five sites in the putative promoter region of NAA60 isoform 2 were analysed. (D) Array-based methylation analysis by Infinium MethylationEPIC BeadChip. The left panel is a heatmap of absolute methylation (β values) for a total of 789 probes located within known imprinted DMRs in the leucocyte samples of the patient (the rightmost lane) as well as 16 control subjects (the other lanes). The nine categories of Δβ values, obtained by the subtraction of the average β value of 16 control leucocyte samples from the β value of patient leucocyte sample, are also shown on the right of the heatmap. (E) qRT-PCR analysis for the leucocytes of the patient and parents. Primer positions are depicted in (A); the primer pair (a) amplifies the transcript of ZNF597 and (b) amplifies NAA60 isoform 1. The graphs represent the mean values (±SD) of three independent experiments, each in triplicate. Statistical significance was determined by unpaired Student’s t test; p<0.05 indicated by *; <0.01 by ** and <0.001 by ***. (F) Allelic expression analysis of ZNF597, NAA60 isoform 1 and 2 determined by direct sequencing of the RT-PCR products encompassing heterozygous SNPs marked with asterisks. DMR, differentially methylated regions; qRT-PCR, quantitative reverse-transcription PCR; Pt, patient; Fa, father; Mo, mother; UPD(16)mat, maternal uniparental disomy of chromosome 16; gDNA, genomic DNA; cDNA, complementary DNA.
Here, we report isolated hypomethylation of the ZNF597:TSS-DMR in a boy with prenatal growth retardation and some dysmorphic features. Even though our patient does not meet the clinical diagnostic criteria for SRS, phenotypic overlap should be noted between our patient and patients with SRS. This is the first case of isolated ZNF597 imprinting defects, which might represent a novel imprinting disorder entity and thus provide new insights into phenotypic spectrum and developmental pathogenesis of imprinting disorders.
Case report
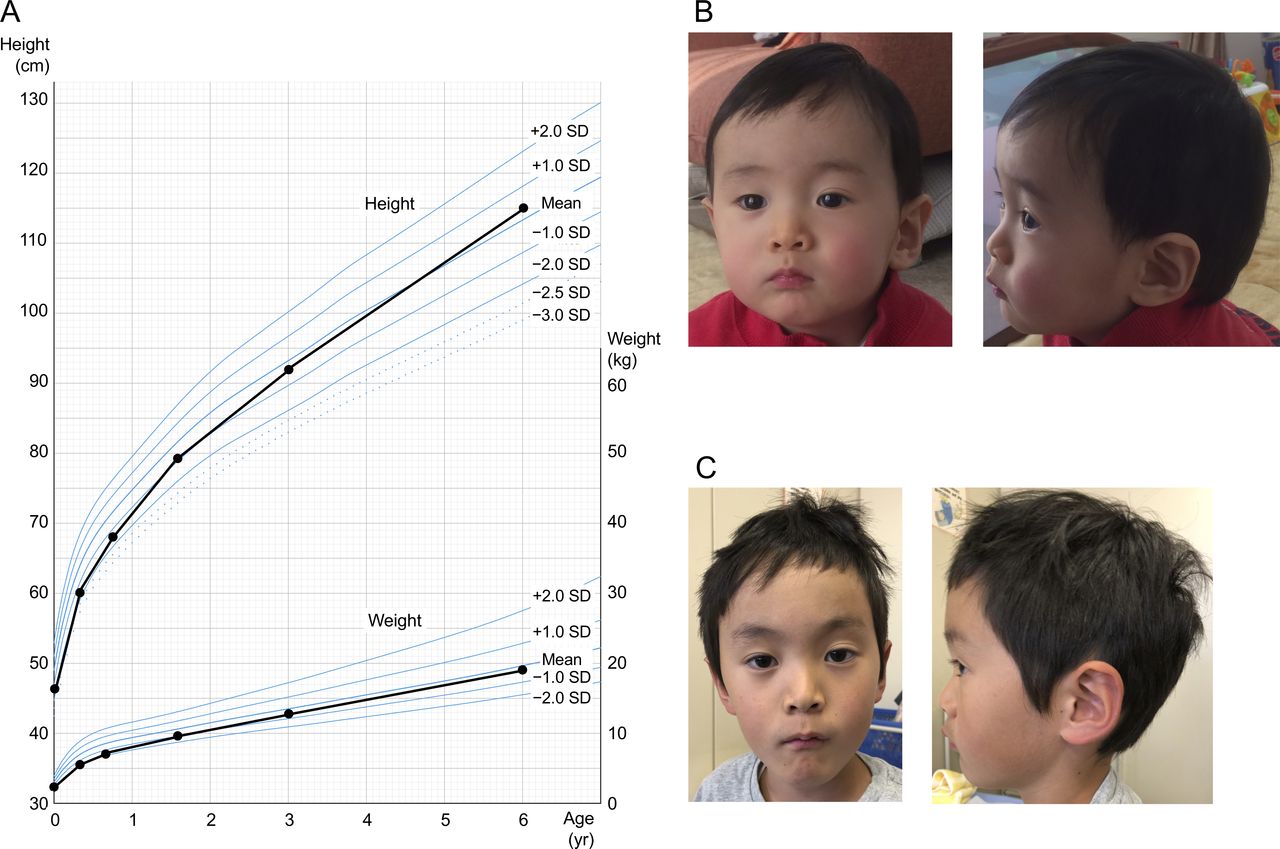
The propositus was identified by a methylation screening cohort of 225 individuals born small for gestational age (SGA), defined as a birth weight below the 10th percentile for gestational age. He was naturally conceived to both 33-year-old non-consanguineous Japanese parents with no remarkable medical history. Intrauterine growth retardation and oligohydramnios had been observed since the third trimester with unknown aetiology and thus careful prenatal follow-up was performed. Serial Doppler ultrasound and cardiotocography studies had indicated reassuring fetal status. He was delivered at 37 weeks of gestation by caesarean section due to fetal growth arrest. His birth weight was 2078 g (−1.71 SD; 4.3 percentile), length was 44.0 cm (−1.43 SD; 7.7 percentile) and head circumference was 32.7 cm (+0.05 SD; 52.0 percentile), which indicated relative macrocephaly at birth. Apgar score was eight at 1 min and nine at 5 min. Placental findings were grossly normal and placental weight was 504 g (−0.18 SD).11 He was admitted to a neonatal intensive care unit due to low birth weight. He required tube feeding for 7 days, but otherwise his clinical course after birth was uneventful. He was discharged from the hospital at 16 days old. He grew up steadily and exhibited catch-up growth (figure 2A). At the age of 3 years, his height was 91.9 cm (−0.42 SD), weight was 12.9 kg (−0.52 SD), head circumference was 50.0 cm (+0.31 SD) and body mass index (BMI) was 15.3 (−0.16 SD). Physical examination revealed mild forehead protrusion at the age of 15 months (figure 2B), but it was less apparent at 6 years (figure 2C). He also had clinodactyly of the right fifth finger, but otherwise manifested no distinctive clinical features associated with known congenital syndromes. His gross psychomotor development was apparently normal. He attends regular class in elementary school. Of note, his 3-year-younger sister was born and has been raised without any growth issues.
{kind=link}
{kind=link}
Clinical findings of the patient. (A) Growth chart indicates that he had prenatal growth retardation, but exhibited catch-up growth. (B,C) Facial features at 15 months (B) and 6 years (C) of age. Note mild protruding forehead was observed at 15 months and less apparent at 6 years of age.
Methods
We describe the detailed methods in online supplementary methods, and primer sequences in online supplementary table 1. In brief, to screen imprinting disorders, we first carried out methylation analysis with pyrosequencing using leucocyte and epithelial buccal cell genomic DNA (Leu-gDNA and Buc-gDNA) for nine DMRs involved in the development of known imprinting disorders, as well as the ZNF597:TSS-DMR, ZNF597:3′ DMR, and putative promoter region of NAA60 gene isoform 2 (NAA60 has two promoters; the promoter of NAA60 gene isoform 1 is located at the ZNF597:TSS-DMR7) (figure 1A). Furthermore, Infinium MethylationEPIC BeadChip (Illumina, San Diego, California, USA) analysis was conducted. Expression levels of ZNF597 and NAA60 were studied using leucocytes by quantitative reverse-transcription polymerase chain reaction (qRT-PCR). To determine parental origins of ZNF597 and NAA60 transcripts, RT-PCR products incorporating exonic heterozygous single nucleotide polymorphisms (SNPs) were Sanger-sequenced. In order to detect UPD(16)mat and hidden mosaic trisomy 16, microsatellite marker analysis was performed. SNP array analysis was conducted to investigate pathogenic CNVs as well as loss of heterozygosity (LOH). Trio whole-exome sequencing (WES) was performed to detect pathogenic variants associated with clinical features such as growth retardation. The genomic region around the ZNF597:TSS-DMR and ZNF597:3′ DMR was directly sequenced to screen for variants that could affect transcription factor binding affinities and cause imprinting defects.12–14
Supplemental material
Supplemental material
Results
Methylation analysis
Pyrosequencing analysis for Leu-gDNA and Buc-gDNA of the patient revealed normomethylation of the nine DMRs related to known imprinting disorders (online supplementary figure 1). Isolated hypomethylation of the ZNF597:TSS-DMR was detected in Leu-gDNA (figure 1C) and Buc-gDNA (online supplementary figure 2C) of the patient, but was not detected in those of the parents. Meanwhile, methylation levels of the ZNF597:3′ DMR were normal in Leu-gDNA of the patient and parents (figure 1C) and Buc-gDNA of the patient (online supplementary figure 2C). The putative promoter region of NAA60 gene isoform 2 were rather hypomethylated in Leu-gDNA of the patient and parents as well as patients with UPD(16)mat analysed as positive controls (figure 1C) and Buc-gDNA of the patient (online supplementary figure 2C), suggesting that this region is not a DMR. Subsequent Infinium MethylationEPIC BeadChip analysis showed that the ZNF597:TSS-DMR was the only DMR that exhibited abnormal hypomethylation in Leu-gDNA (figure 1D; online supplementary table 2) and Buc-gDNA (online supplementary figure 3; online supplementary table 3) of the patient, excluding the possibility of multilocus imprinting disturbance (MLID). Apart from the ZNF597:TSS-DMR, methylation levels of CpG sites across the ZNF597/NAA60 imprinted domain were apparently normal in Leu-gDNA (figure 1B) and Buc-gDNA (online supplementary figure 2B) of the patient. Overall, methylation patterns between Leu-gDNA and Buc-gDNA of the patient were similar across the entire genome, including the ZNF597/NAA60 imprinted domain.
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Expression analysis
qRT-PCR analysis revealed that the expression levels of ZNF597 and NAA60 isoform 1 in the patient were significantly increased (approximately doubled) compared with those in the parents (figure 1E). Genotyping for exonic SNPs in ZNF597 and NAA60 transcripts showed informative in the patient and father, that is, biallelic expression of ZNF597 with leaky transcript from the paternally derived allele in the patient was observed, whereas monoallelic expression was detected in the father, indicating loss of imprinting (LOI) of ZNF597 in the patient (figure 1F). Likewise, NAA60 isoform 1 showed LOI in the patient, but monoallelic expression in the father. Note NAA60 isoform 2 was found to be biallelically expressed both in the patient and father, which was consistent with the previous report.7
UPD and copy number analysis
Microsatellite marker analysis showed biparental inheritance of chromosome 16 in the patient. The area under curve ratios of paternally and maternally derived peaks at each informative locus were similar between Leu-gDNA and Buc-gDNA samples, excluding the possibility of tissue mosaicism (online supplementary figure 4). SNP array analysis did not identify either pathogenic CNVs or LOH regions throughout the entire genome (online supplementary figure 5).
Supplemental material
Supplemental material
Sequencing analysis
Trio-WES did not identify any pathogenic variants associated with growth failure or other distinctive phenotypes (data not shown). Rare variants were also not observed around the sequence of ZNF597:TSS-DMR and ZNF597:3′ DMR.
Discussion
We identified isolated hypomethylation of the ZNF597:TSS-DMR in a 6-year-old boy who showed intrauterine growth retardation accompanied by some dysmorphic features, such as forehead protrusion and clinodactyly of the right fifth finger. Indeed, we successfully demonstrated LOI and subsequent overexpression of ZNF597 in the patient’s leucocytes. Considering that maternal health and obstetric factors affecting fetal development were absent and that other (epi)genetic defects associated with his phenotype such as aberrant methylation at other DMRs or MLID, UPD for chromosome 16 or others, pathogenic sequence variants, CNVs and LOH regions were all excluded, his clinical features were considered to be primarily attributable to hypomethylation of the ZNF597:TSS-DMR. Thus, this is the first report of isolated ZNF597 imprinting defect in humans. Several matters should be pointed out as follows.
First, clinical features of our patient are noteworthy in comparison with those of SRS and UPD(16)mat (online supplementary table 4). Recently, the international consensus statement of SRS recommends adopting the Netchine-Harbison clinical scoring system (NH-CSS) including (1) SGA, (2) postnatal growth failure, (3) relative macrocephaly at birth, (4) protruding forehead, (5) body asymmetry and (6) feeding difficulties and/or low BMI.4 5 Patients meeting four or more of these six criteria receive a diagnosis of SRS. Our patient satisfied two NH-CSS criteria including relative macrocephaly at birth and protruding forehead, thereby indicating that SRS was unlikely. However, these two criteria are the important features that best distinguish SRS from non-SRS SGA.4 Also, he showed prenatal growth failure, the extent to which was not severe enough to meet the SGA definition as birth length and/or weight below −2 SD in NH-CSS. Further, clinodactyly of the fifth finger and tube feeding (until 7 days old) are frequently observed in patients with SRS. Taken together, it is hypothesised that hypomethylation of the ZNF597:TSS-DMR and subsequent overexpression of ZNF597 may develop SRS-specific characteristic features and concomitantly exert potential adverse effects on prenatal growth, thereby leading to phenotypic overlapping of our patient with SRS. In this regard, given that some patients with UPD(16)mat meet the NH-CSS and show SRS phenotype,5 6 ZNF597 overexpression may at least in part comprise the underlying molecular mechanism of developing SRS phenotype in UPD(16)mat. Of note, patients with UPD(16)mat have a non-specific, heterogenous clinical spectrum, such as preterm birth, postnatal growth failure, congenital heart diseases, developmental delay and hypospadias.15 However, these manifestations were not observed in our patient. This might be attributable to the observation that hypomethylation of the ZNF597:TSS-DMR in our patient was less severe than that in patients with UPD(16)mat (figure 1C). Alternatively, considering that recent genome-wide investigations have identified several candidate imprinted genes on chromosome 16 besides ZNF597 and NAA60,16–19 imprinting aberrations of these regions may also cause some specific phenotype of UPD(16)mat, despite the lack of sufficient evidence to date. Also, remarkably, placental hypoplasia was not identified in our patient, implying that ZNF597 overexpression had negative impacts mainly on fetal growth, rather than on placental development, although ZNF597 is expressed in placenta as is the case with most imprinted genes.7
Supplemental material
Second, the causative mechanism by which hypomethylation of the ZNF597:TSS-DMR alone developed is of considerable interest. The ZNF597:TSS-DMR regulates the imprinted maternal expression of both ZNF597 and NAA60. However, the methylation of ZNF597:TSS-DMR is achieved somatically as an sDMR and thus this region is not the ICR of the ZNF597/NAA60 imprinted domain.7 Alternatively, a nearby ZNF597:3′ DMR, located at a 4 kb distance from the 3′ end of ZNF597, is an oocyte-specific methylated gDMR which seems to function as the ICR and regulate the methylation pattern of the ZNF597:TSS-DMR in a hierarchical fashion.8–10 In our patient, the methylation level of the ZNF597:3′ DMR was normal (figure 1B, C and D; online supplementary figures 2B,C and 3). It is thus suggested that failure to acquire substantial methylation of the ZNF597:TSS-DMR (sDMR) alone occurred incidentally during the postimplantation period in our patient, regardless of the methylation status of the upstream ZNF597:3′ DMR (gDMR) being normal. Such a postzygotic event could give rise to hypomethylation with tissue mosaicism, which may also account for the milder phenotype of the patient compared with that of UPD(16)mat, although we confirmed that the ZNF597:TSS-DMR was hypomethylated to a similar extent between Leu-gDNA and Buc-gDNA. Of note, trio-WES, direct sequencing for these two DMRs, and SNP array analysis did not detect any genetic alterations, including pathogenic sequence variants, CNVs or LOH regions which may affect cis-acting elements or trans-acting factors underlying imprinting disorders. Therefore, hypomethylation of the ZNF597:TSS-DMR in our patient is referred to as a primary epimutation that occurred stochastically.
Third, a pathogenic role of increased NAA60 expression awaits further investigations. The NAA60 gene encodes a highly conserved ubiquitous enzyme NAA60 that belongs to a family of enzymes known as N-terminal acetyltransferases.20 In our analysis using leucocytes, the amount of relative expression of NAA60 isoform 1 in the patient was significantly increased, similarly to that of ZNF597 (figure 1E). Hypomethylation of the ZNF597:TSS-DMR could contribute to LOI of NAA60 isoform 1 as well as ZNF597, which yielded increased expression from the paternally derived allele that was originally supposed to be imprinted. The RT-PCR assay to determine the allele-specific expression using the heterozygous SNP rs1137454 demonstrated this perturbed imprinting. The possibility that overexpression of NAA60, especially isoform 1, would also exert potential effects on the phenotype of our patient should be considered; however, the biological role of NAA60 involving human disease remains largely unknown thus far.
In summary, we identified isolated hypomethylation of the ZNF597:TSS-DMR as a primary epimutation in a boy with prenatal growth retardation and some dysmorphic features reminiscent of SRS. Our case raises the possibility that ZNF597 overexpression may underly the development of SRS phenotype observed in UPD(16)mat. In particular, given that our patient exhibited mild intrauterine growth retardation and catch-up growth, it may be worth considering molecular testing for the ZNF597/NAA60 domain in patients with clinical suspicion of SRS, regardless of the severity of growth restriction. This is the first report of isolated ZNF597 imprinting defect; however, we have identified only one patient with this epimutation in a methylation screening cohort of 225 individuals born SGA, thereby providing limited evidence. Thus, further investigations and case series are needed to establish a novel imprinting disorder entity involving the ZNF597/NAA60 domain.
Ethics statements
Ethics approval
This study was approved by the Institutional Review Board Committees at the National Hospital Organisation (reference number: H30-0125001) and at the National Center for Child Health and Development (reference number: 1814).
Acknowledgments
We are grateful to the patient and family members for participating in this study. We also thank the medical editor from the Division of Education for Clinical Research at the National Center for Child Health and Development for editing this manuscript.
References
Footnotes
Contributors KY designed and coordinated the study and drafted the manuscript. KY, TI, MN, AN and KM performed molecular analysis. YS, TN and HY provided detailed clinical data and materials for molecular analysis. KK, HF and KE provided normal control samples. KN, KH, TM, TO and MK supervised the study. All authors read and reviewed the manuscript.
Funding This work was supported by a Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (16H05362), a Grant-in-Aid for Clinical Research from the National Hospital Organization (H29-NHO(SEIIKU)−01), a Grant from the Takeda Science Foundation, a Grant from the Yamaguchi Endocrine Research Foundation, a Grant from the Foundation for Growth Science, a Grant from the Japanese Society for Pediatric Endocrinology and a donation from the Enomoto Children’s Clinic.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.