Article Text

Abstract
Background Dysfunction of histone methyltransferases and chromatin modifiers has been implicated in complex neurodevelopmental syndromes and cancers. SETD1B encodes a lysine-specific methyltransferase that assists in transcriptional activation of genes by depositing H3K4 methyl marks. Previous reports of patients with rare variants in SETD1B describe a distinctive phenotype that includes seizures, global developmental delay and intellectual disability.
Methods Two of the patients described herein were identified via genome-wide and exome-wide testing, with microarray and research-based exome, through the CAUSES (Clinical Assessment of the Utility of Sequencing and Evaluation as a Service) Research Clinic at the University of British Columbia. The third Vancouver patient had clinical trio exome sequencing through Blueprint Genetics. The fourth patient underwent singleton exome sequencing in Nantes, with subsequent recruitment to this cohort through GeneMatcher.
Results Here we present clinical reports of four patients with rare coding variants in SETD1B that demonstrate a shared phenotype, including intellectual disability, language delay, conserved musculoskeletal findings and seizures that may be treatment-refractory. We include supporting evidence from next-generation sequencing among a cohort of paediatric patients with epilepsy.
Conclusion Rare coding variants in SETD1B can cause a diagnosable syndrome and could contribute as a risk factor for epilepsy, autism and other neurodevelopmental phenotypes. In the long term, some patients may also be at increased risk for cancers and other complex diseases. Thus, longitudinal studies are required to further elucidate the precise role of SETD1B in neurodevelopmental disorders and other systemic disease.
- genetics
- clinical genetics
- epilepsy and seizures
- molecular genetics
Statistics from Altmetric.com
Introduction
Protein-altering variants in histone methyltransferases, and in chromatin modifiers more generally, have been implicated in complex neurodevelopmental syndromes and in cancers. Neurodevelopmental syndromes associated with these variants are typically dominant and occur sporadically due to de novo pathogenic mutations. Parent-to-child transmission of these variants occurs rarely, often due to parents having somatic or gonadal mosaicism for the mutation. The presence of three independent families in which the same rare phenotype associates with different de novo mutations in the same gene allows causal inference for those genotype–phenotype correlations. This is especially true when perturbation of the general biological process has, in similar contexts, been shown reproducibly to result in similar phenotypes.
Individual case reports have appeared in the literature describing neurodevelopmental delays associated with rare coding variants in SETD1B, and with hemizygosity for SETD1B due to CNVs. Here we show that pathogenic variants within SETD1B contribute to a conserved phenotype that includes intellectual disability and childhood-onset, treatment-refractory seizures.
SETD1B encodes the protein SET domain-containing protein 1B, a lysine-specific methyltransferase involved in histone methylation.1 Working in close association with four other subunits, SETD1B forms the catalytic component of a COMPASS (Complex Proteins Associated with Set1) histone modifier, methylating histone H3 at Lys4 up to three times.1 2 This trimethylated histone mark is believed to assist in the transcriptional activation of nearby genes. Thus, SETD1B is an epigenetic writer that modifies chromatin structure, with downstream effects on gene expression.2
Previous reports by Hiraide et al and Den et al described four Japanese patients, aged 1, 7, 10 and 34 years, whose phenotypes included intellectual disability, seizures and autistic features.3–5 All of these patients also had craniofacial, musculoskeletal and cutaneous abnormalities.
Published reports of patients with microdeletions encompassing SETD1B document similar phenotypes. Three affected patients had seizures, and one had moderate-severe intellectual disability.6–8 Additional findings included receptive and expressive language disorders as well as motor delays.6–8 Most recently, Krzyzewska et al 9 described a specific DNA hypermethylation signature believed to be associated with SETD1B loss of function, which was used to reclassify two SETD1B variants of uncertain significance (VUS) as pathogenic.
Here we present detailed descriptions of four additional patients, along with rare SETD1B alleles identified in a cohort of patients with epilepsy. The de novo variants in SETD1B are as follows: a splice-site variant c.5589+1G>A (p.?), nonsense variants c.2932C>T: p.(Gln978Ter) and c.3964C>T p.(Gln1322Ter), and a missense variant c.5833T>C p.(Phe1945Leu). Our replication study consisted of comparing the number of rare coding variants in SETD1B identified in a sequentially recruited cohort of 207 children and adults with childhood-onset epilepsy, with the number of SETD1B variants of similar or lower minor allele frequency (MAF) found among a heterogeneous cohort of patients with neurological disorders excluding epilepsy. In the context of previously reported cases, these patients confirm the implication of rare SETD1B variants in paediatric epilepsy and confirm a shared phenotype produced by some de novo, highly penetrant variants. This phenotype includes intellectual disability, language delay, conserved musculoskeletal findings and seizures that may be treatment-refractory.
Methods
Individuals underwent whole genome or exome sequencing through several studies and diagnostic laboratories, including the CAUSES study (Clinical Assessment of the Utility of Sequencing and Evaluation as a Service), Blueprint Genetics, the Service de Génétique Médicale in Nantes and EPGEN (Epilepsy Genetics Study).10–12 Analysis methods for these services have been detailed previously.10 11 In brief, individuals 1 and 2 had trio genome sequencing through CAUSES, a study assessing the application of exome sequencing as a hospital-based service. Individual 3 had clinical trio exome sequencing (Whole Exome Family Plus), through Blueprint Genetics in Helsinki, Finland. Individual 4 underwent clinical exome sequencing with variant confirmation via Sanger sequencing in Nantes, France.
Given the emerging association between rare coding SETD1B variants and epilepsy, variant lists derived from Vancouver’s EPGEN study database were queried as a replication set.11 Crude ORs were estimated in two ways: in comparison with controls affected by non-epileptic neurogenetic disorders and in comparison with the control subset of the Genome Aggregation Database (gnomAD). These latter samples exclude cases from common disease case/control studies (https://macarthurlab.org/2018/10/17/gnomad-v2-1/). For reporting allele counts (for both gnomAD controls and the total gnomAD population), the number used is the median number of subjects with genotyping at all points which passed quality control metrics across the entirety of the SETD1B region.
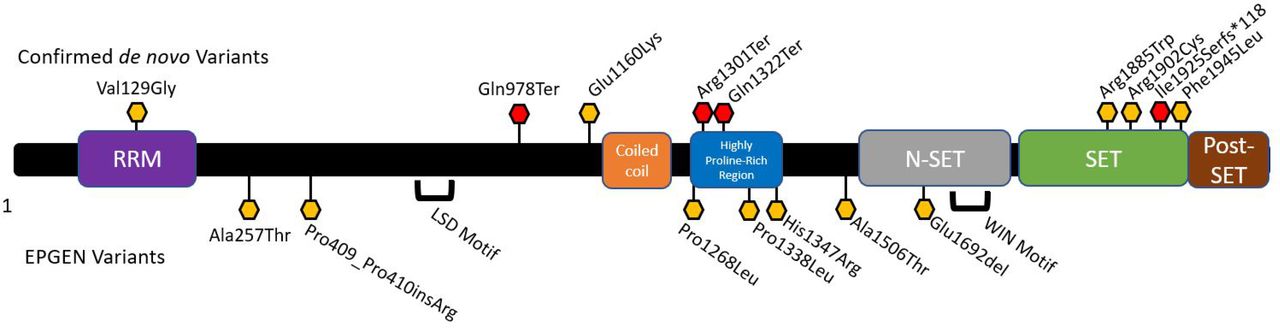
We attempted to synthesise the existing experimental data on human SETD1B by visual inspection of the domains listed as its most recent iteration in the UniProt Knowledge Base (UniProtKB entry Q9UPS6), followed by manual curation of the references cited there.13 Experimental data and evolutionary conservation support the existence of an RNA recognition motif, an lysine-specific histone demethylase (LSD) motif, a coiled-coil domain, an N-SET domain containing a WDR5 interacting motif, a SET domain and a post-SET domain (figure 1, online supplementary figure S1 and table S1).1 13–17 Domain boundaries are listed in online supplementary table S1. We then tallied domain-specific allele frequencies observed in the EPGEN database (online supplementary table S2), among neurogenetic patients without epilepsy, and in gnomAD controls (online supplementary table S3). We summed variant counts across the specified domain for each variant type (missense single nucleotide variant, splice site, total). OR calculations are based off of the total number of variants in each subclass, and on either the participant total (for the EPGEN and non-EPGEN neurological cohorts) or the median number of individuals as counted across the entire gene (control gnomAD population).
Supplemental material
Supplemental material

Schematic of the human SETD1B protein. This schematic is annotated with putative domains and the rare variants described in this report. Red hexagons indicate chain-terminating variants, while yellow hexagons indicate missense or insertion variants. Confirmed de novo variants are listed above, while variants from the EPGEN cohort (n=207 paediatric epilepsy cases) are listed below. EPGEN, Epilepsy Genetics Study; N-SET, component N motif for Complex Proteins Associated with Set1p (~residues 1668–1821); HPRR, highly proline-rich region (~residues 1284-1364); Post-SET, cysteine-rich motif that follows the SET domain (~residues 1950–1966); RRM, RNA recognition motif (approximate boundary residues 93–181); SET, Su(var)3–9-enhancer-of-zeste-trithorax domain (~residues 1821–1944); WIN, WDR5 interacting motif.
To further investigate the possibility that rare pLoF variants observed in paediatric-onset disease had different properties than rare variants observed among gnomAD participants, we manually curated the 14 variants annotated as pLoF (in gnomAD entry V.2.1.1) and compared these with the SETD1B variants downloadable from the gnomAD participants listed as controls. Three-dimensional modelling (online supplementary figure S2) was based on the crystal structure of the MLL1 SET domain as described previously.18–20 Tissue-specific expression (online supplementary figure S3) was obtained from GTEx Analysis Release V.8 (gtexportal.org) by searching for the HUGO Gene Nomenclature Committee (HGNC) gene symbol SETD1B.21
Supplemental material
Supplemental material
Clinical reports
Individual 1
A 7-year-10-month-old boy presented to the Medical Genetics with a history of seizures, global developmental delay and moderate intellectual disability.
He had eyelid myoclonia at 2 ½ years old, and at 3 ½ years he manifested absence seizures after the eyelid myoclonia. Seizures lasted 1–2 s and consisted of eye-rolling and eyelid flickering. They initially occurred several times per week before increasing to over 30 seizures per day. EEG showed synchronous spike and wave activity. CT was normal and MRI at 5 years showed a somewhat bulky corpus callosum and multiple tiny foci of high Fluid attenuation inversion recovery (FLAIR)/T2 signals in the white matter, mostly in the frontal regions. These foci were non-specific; differential diagnosis at that time included a syndromic disorder, previous non-specific injury and previous infection.
Neurological evaluation documented treatment-resistant eyelid myoclonia with absence seizures, attention deficit hyperactivity disorder (ADHD) with moderate intellectual disability, and expressive and receptive language delays. During a recent febrile illness, there had also been several brief episodes of slight jerking and stiffening of his arms. Following illness resolution, the limb movements had gradually resolved, but his eye-rolling seizures increased in frequency.
Valproate, clobazam, lamotrigine, levetiracetam, Biphentin and oral cannabis products were trialled with little success. Ethosuximide provided some seizure control, although he continued to have dozens of seizures per day. He was also taking dextroamphetamine for ADHD, vitamin supplements and omega-3 oil.
He was born via induction at 41 weeks and 4 days, following an unremarkable pregnancy. Birth weight was 3402 g (+0.41 SD). He required neonatal resuscitation, but the newborn period was otherwise uncomplicated. Early milestones were delayed. He sat at 9 months, walked at 15 months and pedalled a tricycle at 3 ½ years. He could dress himself at 4 ½ years. He used single words at 2 years and formed sentences of three to four words at age 3 ½ years. By 4 ½ years, he was speaking sentences of six to seven words. Receptive language was also impaired, and he had challenges following directions. At age 4 ½, he did not know body parts but could name several colours.
Physical examination revealed height and weight within −0.02 SD and +1.95 SD of the mean, respectively. He did not have dysmorphic features; however, he did have a small midline pit below the nasal septum, where the septum and philtrum meet. Cutaneous findings included a 2 cm hypopigmented patch below his right scapula. Neurological examination was unremarkable.
This patient was the only child of non-consanguineous parents, and the family history was unremarkable. Work-up for small molecule metabolic diseases was negative, and formal assessment did not suggest autistic features. Fragile X testing and clinical microarray were also negative. Trio exome sequencing revealed a de novo heterozygous variant of unknown significance in SETD1B (c.5589+1G>A(p.?)). This specific variant has not been previously reported in population databases and is predicted to affect splicing.
Individual 2
This 14-year-8-month-old girl presented to genetics for further work-up of epilepsy, autism and developmental delay. Her first seizure occurred at age 11 years, followed by infrequent tonic-clonic seizures approximately twice a year. Her seizures included right-sided facial twitching and blinking, and were worse on waking. At 12 years old, she was admitted to hospital for a seizure likely secondary to medication refusal. EEG demonstrated atypical spike waves, fragmentary spike waves and bifrontal slowing following eye-blinking. MRI appeared normal.
Valproate led to increased ammonia and low platelets. Clobazam was also trialled, but led to weight gain and severe behavioural challenges. She was globally delayed and dressed and bathed independently by age 15. She had a wide vocabulary, but only communicated in short phrases (in English, Thai and Mandarin). At age 19 years, she was seizure-free on topiramate and ethosuximide after clobazam was weaned from her regimen.
The prenatal course was unremarkable, and she was born at 41 weeks by vacuum assistance (birth weight not recorded). She was the eldest child of non-consanguineous Thai parents. She was diagnosed with autism spectrum disorder during early childhood. She also had an otolaryngology referral for enlarged tonsils and loud snoring; however, she did not require any treatment for these symptoms. She had normal ECG, and patent ductus arteriosus that was too small to require surgery but still requires endocarditis prophylaxis for dental work. At age 12, she was diagnosed with anovulatory menstrual cycles and menorrhagia, and was treated with progestin therapy.
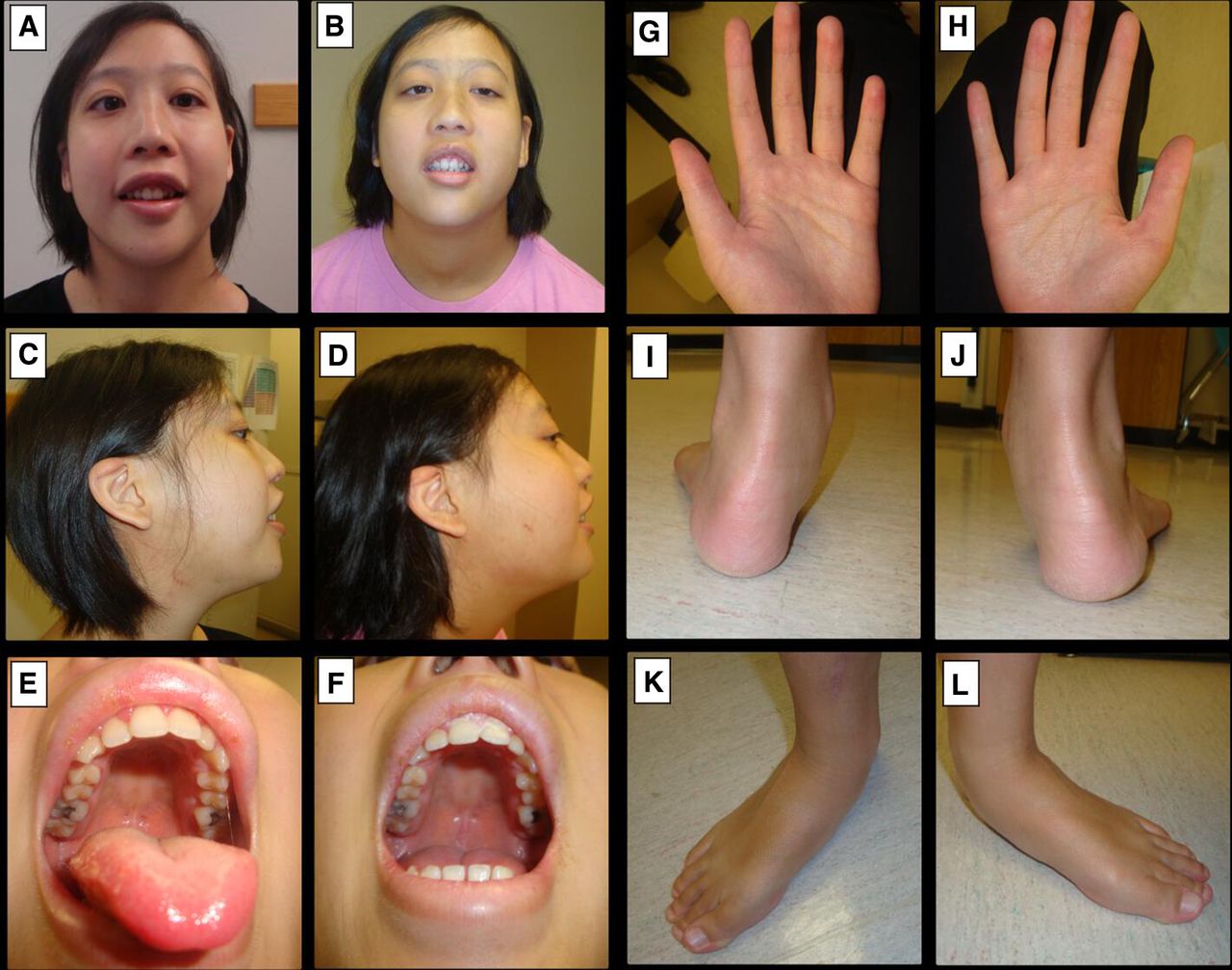
At her initial presentation to the Medical Genetics, her head circumference was at +0 SD, her height was at +1.8 SD and her weight at +1.6 SD. She did not have any facial dysmorphisms; however, she did have sparse hair. She had mild joint laxity at the metacarpophalangeal joints and long tapering fingers (figure 2). She had lumbar lordosis and a right rib prominence, with the right shoulder higher than the left. She had pigmentation changes and scars related to previous eczema.

{kind=link}
{kind=link}
Features of individual 2, heterozygous for p.(Gln1322Ter). Facial features for individual 2 at age 14 years and 8 months (A, C, E) and at age 19 years and 6 months (B, D, F). She had full cheeks and full lips, but was not felt to be dysmorphic on examination. She had a normal hairline, but sparse hair. Her palate was normal, without a cleft or groove. She was evaluated by otolaryngology for enlarged tonsils and snoring, but did not require surgery. Her right ear had an overfolded antihelix. Hands and feet of individual 2 at age 19 years and 6 months (G–L). She had long, tapering fingers (G, H) and metacarpophalangeal laxity. She had tibiotalar eversion (I, J) and pes planus, and her second digit was longer than the first (K, L).
She had a tongue tremor with slightly decreased muscle bulk on the right side of the tongue. She also had spontaneous periodic shaking of the hands medially and laterally. Her gait suggested tight heel cords, with some circumduction.
At age 19 years and 7 months, her height was 172 cm (+1.4 SD, but mid-parental height was 161 cm) and her weight was +0.9 SD. Head circumference was 55 cm (+0.63 SD).
She had two healthy siblings. A paternal uncle had passed away of lymphoma at age 36.
Metabolic testing was unremarkable, and trio exome sequencing at age 18 years revealed heterozygosity for a de novo c.3964C>T (p.Gln1322Ter) SETD1B variant (see figure 1). This variant is predicted to create a premature stop codon leading to nonsense-mediated mRNA decay.
Individual 3
This patient was 3 years and 6 months old at last follow-up. His seizures manifested at approximately 2 ½ years, as staring spells with occasional head droop. He had subtle dysmorphic features (downslanting palpebral fissures, small ears, mild mid-facial hypoplasia, small nose, smooth philtrum, thin upper lip, narrow chin), mild fifth finger clinodactyly and persistent fingertip pads.
His seizures were initially described as a ‘sudden behavioural arrest’, lasting about 1–2 s and consisting of staring and a head droop near the end of the event. These seizures occurred several times per hour, for approximately a year. There were no other seizure types. His EEG was diagnostic for photosensitive generalised absence seizures with a probable myoclonic component. At most recent evaluation, he was being treated with valproic acid.
This patient was born at term following an unremarkable pregnancy to non-consanguineous parents; family history was unremarkable. He weighed 3062 g (−0.60 SD). He had global developmental delay. He was late to sit unassisted, walked at 18–20 months, and had a modified pincer grip at 3 years. He spoke his first words at 27 months, and did not use pointing or typical gestures to communicate. By age 3 years, he spoke short sentences that were 50% intelligible. He was able to draw simple shapes, but was not potty-trained. He had a social smile and was interested in others. He would occasionally stare at toys or play with them in a repetitive manner.
Additionally, he also had oral hypotonia diagnosed at 2 weeks, associated with poor weight gain. With parental education and feeding modifications, he gained weight and had no further feeding issues. He required orthoses. Right inguinal hernia was repaired at 3 years and 10 months. Formal testing diagnosed autism spectrum disorder at 3 years and 10 months. MRI showed non-specific scattered foci of subcortical T2 hypodensity.
At the time of his last assessment in the Medical Genetics, his height and weight were at +1.0 SD and +0.6 SD of the mean, respectively. His head circumference was at −1.3 SD below the mean. He had subtle dysmorphic features, but the remainder of his examination was normal.
Metabolic testing and fragile X were unremarkable. Exome sequencing found a de novo heterozygous missense variant in SETD1B at c.5833T>P p.(Phe1945Leu).
Individual 4
This patient was 12 years and 3 months old at last follow-up. He had a history of epilepsy and moderate intellectual disability.
Seizure onset was at 2 years and 6 months, and included ocular revulsion and absences. He began therapy with lamotrigine at 3 years and 6 months. He later developed severe generalised epilepsy and myoclonia. There is no EEG available for this patient, but he had normal MRI.
He was born via caesarean delivery at term following an unremarkable pregnancy. His birth weight was 3930 g (+1.13 SD). He had neonatal feeding difficulties and a developmental delay. He walked at 16 months, and at 12 years old he could not read or write. He did not have a speech delay. He had moderate intellectual disability and was occasionally aggressive.
Last follow-up was at 12 years and 2 months, when his height and weight were −0.29 SD and +2.51 SD of the mean, respectively. Head circumference was at +1.65 SD.
Singleton exome sequencing with Sanger validation in parents revealed an apparently de novo nonsense variant in SETD1B at c.2932C>T: p.(Gln978Ter).
Assessment of rare SETD1B variant burden in paediatric-onset epilepsy
To investigate the potential contribution to epilepsy in paediatric tertiary care, rare variants (MAF <0.01) in SETD1B were sought among 207 affected children enrolled in the EPGEN study at the British Columbia Children’s Hospital, compared with 3002 cases of individuals with neurological phenotypes excluding seizure disorders.11 EPGEN variant frequencies were also compared with rare alleles in the gnomAD ‘controls’ subset (defined as the participants who remained after exclusion of those identified as ‘cases’ in participating case–control data sets). EPGEN-derived SETD1B variants are provided in online supplementary table S2. Neither Sanger validation nor parental testing was done for this group of variants, although next-generation sequencing and bioinformatic analyses used the same protocols. For the calculation of OR the NeuroSeq cohorts had been annotated with respect to NM_015048.1. Full reannotation of the NeuroSeq database to NM_001353345.1 is not feasible at this time. The original annotations were retained for comparison with gnomAD variants (online supplementary table S3), but reannotation would be unlikely to change the ORs significantly. The variants themselves are ranked as VUS, so the ORs are suggestive of what might apply to truly pathogenic variants. Online supplementary table S4 shows OR across the whole coding region, as well as computed on a per-domain basis where possible. Online supplementary table S5 shows OR computed using increasingly conservative thresholds set for rarer alleles (MAF of 0.005 and below). Because of the smaller size of the EPGEN cohort relative to the gnomAD controls, few ultra-rare alleles of MAF <0.0001 were detected in EPGEN, which may help to explain the apparent paradoxical decrease in the calculated OR with decreasing allele frequency.
Patients with paediatric epilepsy are almost twice as likely to have a rare SETD1B VUS as are gnomAD control participants (~2% prevalence vs ~1% prevalence, formal OR 1.8). When cases with paediatric epilepsy are selected out of a heterogeneous cohort of neurological disorders, the OR of a SETD1B VUS increases to 5.9. Thus, sequestration of cases with childhood-onset epilepsy moderately sequesters rare variants in SETD1B as well. Most of the increase in odds seems to be driven by a cluster of missense variants in a region of SETD1B that is rich in prolines, which we have defined by visual inspection as extending from residues 1284–1364 (online supplementary table S1, figure S1). UniProtKB lists a region of compositional bias rich in prolines from positions 366–1671 in SETD1B.13 However, visual inspection of the distribution of rare alleles from the EPGEN cohort and published reports suggested a clustering of missense variants within a subregion that spans approximately residues 1284–1364, within which 40% of the residues (32 out of 80) are prolines. This highly proline-rich region may face steric constraints that render missense variants more damaging here than elsewhere. In general, missense SNVs and splice-site SNVs are over-represented in the EPGEN cohort relative to gnomAD controls.
Splice-site variants may have effects that vary by tissue, and missense variants may cause varying degrees of loss-of-function (LoF), gain-of-function, may create a novel function, or be neutral. Thus, OR comparisons like the above are intrinsically poorly powered and crude. Online supplementary table S6 shows the pLoF variants in SETD1B from the gnomAD cohort. Of the 14 pLoF variants annotated in gnomAD V.2.1.1, 8 were flagged as being of dubious quality. Four of the remaining variants were not seen among gnomAD controls, which suggests that they are present among the cases in the case–control studies that make up gnomAD. Manual curation of online supplementary table S6 yielded an unexpected observation: out of the six ultra-rare high-confidence pLoF variants affecting SETD1B, four were observed in the Finnish population. The c.2715+8_2715+15delGGTGGGTG variant is annotated in a single Finnish individual and appears in the gnomAD control data set. The p.Trp29Ter, p.Ala1463ProfsTer10 and c.5598+1G>C variants all appear once only in Finnish individuals not annotated as controls. Since all of these variants passed similar quality control metrics in the gnomAD protocol, a crude OR of 4.13 can be calculated for rare SETD1B pLoF variants within the Finnish population. Because Finland has a unique population history, and because the ‘cases’ within gnomAD were ascertained as having a variety of different adult-onset diseases (including type 2 diabetes, cardiovascular disease and schizophrenia), the generalisability of such an OR is difficult to assess.
A variant we annotated as NM_015048:c.1226_1226delinsCCCG was seen twice in the EPGEN cohort, and is predicted to insert an arginine between prolines 409 and 410 (online supplementary figure S1, table S2). In one case, the variant was predicted to be de novo on the basis of its presence in the proband and absence from parental exomes (minimum 20× coverage at that site in both), and in a second case the parents were unavailable and it was detected in the proband alone. This variant was also observed once in a non-epilepsy neurogenetic case. None of the three cases has been Sanger-verified. Assuming it is real and not a rare protocol-specific false discovery places the global OR for ultra-rare SETD1B alleles at 1.54, and assuming it is an artefact would place the global OR for ultra-rare SETD1B alleles at 1.01 (online supplementary table S5).
Three-dimensional modelling of the SET domain (online supplementary figure S2) was based on the published structure of the MLL1 SET domain.18 The de novo missense variants associated with epilepsy (from individual 3 and Hiraide et al 3) are predicted to lie within the SET domain’s histone-binding pocket. Modelling of missense VUS from the gnomAD cohort that lie within the SET domain predicts that the side chains of these residues lie outside of the histone-binding pocket, which is consistent with the lower a priori expectation of functional consequences for these variants.
Discussion
Comparison of patients with isolated SETD1B mutations
There are several similarities between our cases and the three patients previously reported by Hiraide et al 3 and Den et al.4 5 Most patients were diagnosed with seizures prior to 4 years of age (table 1). All eight patients also have varying degrees of intellectual disability, with six having language delays. Many patients also failed multiple seizure medications, and six of eight patients had autism spectrum disorder. These data show that de novo or apparently de novo SETD1B missense and pLoF variants are likely to perturb neurodevelopment significantly.
Phenotypes of patients with SETD1B variants.
Phenotypic comparison with microdeletion patients
To date there have been seven reports of patients harbouring deletions that encompass SETD1B, the first appearing in 1999. These patients share several phenotypic traits with our cases. Most notably, three of these seven patients have seizure disorders with abnormal EEG findings. Two patients had intellectual disability, and four patients had significant language delays. Two of these patients were diagnosed with autism spectrum disorders.
These patients also tended to have similar craniofacial dysmorphisms, including full cheeks, nasal abnormalities, macroglossia and tapering fingers.6–8 22 23 More than half (four of seven patients) also had dermatological abnormalities, including café au lait spots, eczema and ichthyotic skin, and hyperpigmentation and hypopigmentation abnormalities.8 22–24
However, many earlier reports have limited phenotype descriptions (five of seven reports did not comment on intellectual disability; two of seven did not comment on seizures). It is therefore possible that some phenotypic traits are under-reported in the existing literature.
Interestingly, one patient’s microdeletion was inherited from an apparently unaffected father.8 This raises questions about penetrance and possible maternal imprinting associated with the 12q24 chromosome region.8
Frequencies of clinical features reported to date among patients with isolated SETD1B variants and those with microdeletions are provided in table 2.
Comparison of patients with isolated SETD1B variants and SETD1B microdeletion
SETD1B and malignancy
Variants in H3K4 methyltransferases, including variants in SETD1B, have been associated with several cancer processes, including leukaemia and oesophageal squamous cell carcinoma.15 25 26 In endometrial carcinoma, SETD1B may also have a role in predicting myometrial invasion and therefore prognosis of endometrial carcinoma.27 Although the significance of SETD1B variants in tumours remains unclear, functional studies suggest that disruption of histone regulation plays a critical role in disease pathogenesis.26–29
Interestingly, several previously reported patients have also been diagnosed with malignancies. The male patient described by Hiraide et al 3 was diagnosed with tubular adenocarcinoma of the sigmoid colon at age 30 years. A female microdeletion patient reported by Qiao et al 22 was also diagnosed with a T cell skin lymphoma during childhood.
The four individuals described in this report did not undergo any additional screening for malignancy risk. Previously reported malignancies range in type and organ system, and additional screening or biomarker testing would have low sensitivity, with a strong possibility of test results of unclear significance (ie, mild elevations of biomarkers, and/or minor congenital anomalies that were difficult to distinguish from benign or preneoplastic growths). At this time, more functional data and larger patient cohorts are needed in order to further elucidate any possible role for constitutional SETD1B variants in the development of primary malignancies.
Population genetics of rare coding variants in SETD1B
Rare missense and LoF variants are relatively under-represented in the gnomAD database.30 Predicted LoF variants in SETD1B are exceedingly rare, with only four splice donor variants, one stop gain and one frameshift catalogued there once variants of dubious quality are removed (total: 6 pLoF alleles out of 141 456 participants). In this respect, it is even more constrained than SETD1A (2 splice acceptor, 3 stop gain and 7 frameshift variants, thus 15 pLoF alleles). Since de novo LoF and frameshift mutations in SETD1B cause epilepsy, the relative absence of frameshift variants from a population of adults without paediatric-onset disease is unsurprising.5 Furthermore, epigenome-wide profiling among patients with de novo pathogenic SETD1B alleles, including some deletion patients, has found a hypermethylation signature associated with these mutations.9 This finding shows downstream consequences at the DNA level of perturbations of H3K4 methylation and offers an intriguing possibility to discern rare VUS from pathogenic alleles via DNA methylation evidence.
The de novo missense variants reported by Hiraide et al 3 4 in 2018 and 2019 are predicted to lie within the histone-binding pocket of the SET domain, as is individual 3’s SETD1B variant (online supplementary figure S2). Several rare missense variants in the SET domain are reported among gnomAD controls, although these are generally predicted to lie outside of the histone-binding pocket. The concordance of the patient phenotypes with those of patients with pLoF variants, along with the predicted functionally sensitive location of these missense variants, suggests that these are also LoF missense variants.
Recently, rare variants in SET domain-containing 1A (SETD1A) have been associated with schizophrenia and other complex neurodevelopmental disorders.31 Mutations in SETD1A have also been implicated in early-onset seizure disorders; both genes have broadly overlapping but distinct expression profiles (online supplementary figure S3, based on GTEx data).32 SETD1A is also a COMPASS H3K4 lysine methyltransferase and is closely associated with chromatin remodelling.31 32 Functional studies have suggested that these mutations may affect normal synaptic development, thus supporting the hypothesis that dysregulation at the level of H3K4 methylation can affect chromatin remodelling negatively, with downstream consequences for neuronal function.
Conclusion
Taken together, our de novo cases document an emerging phenotype associated with SETD1B disruption, which includes global developmental delay, intellectual disability, autism, subtle congenital anomalies without a strong facial resemblance between patients and difficult-to-control seizures. Our study is limited to genetic data and to correlations of rare phenotypes with de novo mutations; functional studies to compare H3K4 methylation and other post-translational modifications in relevant tissues would help to identify the transcriptomic perturbations that result from SETD1B mutations of large functional effect. Rare coding variants in SETD1B are strongly enriched among cases of paediatric-onset epilepsy in comparison with non-epileptic controls based on crude ORs, and rare pLoF SETD1B variants appear to be somewhat enriched among gnomAD cases of Finnish descent relative to controls. Notably, rare LoF variants in similar chromatin modifiers have been proven to confer risk for schizophrenia and other neurodevelopmental disorders (eg, SETD1A), a finding which anchors perturbation of H3K4 methylation as a biological process that confers risk for these complex neurodevelopmental phenotypes. Rare functional variants in SETD1B could plausibly contribute as a risk factor for autism and other neurodevelopmental phenotypes in the general population without paediatric-onset epilepsy. The long-term risks for cancers and for other common, complex diseases among individuals with SETD1B-related neurodevelopmental disorders are unknown and merit further longitudinal study.
Acknowledgments
The bioinformatic pipeline used in part of the CAUSES study was developed in the laboratory of Wyeth Wasserman.
References
Footnotes
Collaborators CAUSES study investigators include Shelin Adam, Nick Dragojlovic, Christèle du Souich, Alison M Elliott, Anna Lehman, Larry Lynd, Jill Mwenifumbo, Tanya N Nelson, Clara van Karnebeek and Jan M Friedman (PI). EPGEN study investigators include Shelin Adam, Cyrus Boelman, Corneliu Bolbocean, Sarah E Buerki, Tara Candido, Patrice Eydoux, Daniel M Evans, William T Gibson, Gabriella Horvath, Linda Huh, Tanya N Nelson, Graham Sinclair, Tamsin Tarling, Eric B Toyota, Katelin N Townsend, Margot I Van Allen, Clara van Karnebeek and Suzanne Vercauteren.
Contributors As the first author, AR was involved in manuscript writing, data interpretation and creating patient figures. DE and MF performed the statistical analysis of the data and contributed significantly to drafting the manuscript. HG, MM, BI, BC, MD and MC all contributed patient cases and contributed to manuscript revisions. Additionally, MD and MC assisted in initial patient recruitment to the EPGEN study and provided neurological expertise in describing the cases. JM and CvK are investigators in the CAUSES study and contributed to drafting and revision of the manuscript. JA and SJMJ contributed their expertise regarding modelling and interpretation of the SETD1B protein, as well as the creation of figures detailing domain characteristics. As the last author, WTG assisted in data acquisition, interpretation, manuscript writing, figure and table preparation, and overseeing project development. All authors have agreed to be responsible for the accuracy and integrity of this manuscript.
Funding The CAUSES study was made possible by a $3 million donation from Mining for Miracles through the BC Children’s Hospital Foundation and is supported by Genome British Columbia, the Provincial Health Services Authority, BC Children’s and Women's Hospitals, the Provincial Medical Genetics Program, BC Children's Hospital Pathology and Laboratory Medicine, and the University of British Columbia. This study was funded by BC Children's Hospital Intramural Funds, Canadian Institutes of Health Research (project grant: PJT-148695, PJT-148830). WTG's salary is supported by the BC Children's Hospital Foundation through their Investigator Grant Award Program (IGAP).
Competing interests WTG reports grants from the Canadian Institutes of Health Research, grants from Heart and Stroke Foundation of Canada, and grants from the Rare Disease Foundation. DE is named in a disclosure to the University of British Columbia regarding the creation of software for the annotation and analysis of human genetic variation in the context of disease. Some of the data analyses performed in this paper were done using the data stored and annotated by this software, which is available under a licensing agreement to third parties. MD has received research support from the Rare Disease Foundation and the Alva Foundation.
Patient consent for publication Parental/guardian consent obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information. Data represented deidentified participant data, available from the CAUSES and EPGEN studies (University of British Columbia).