Article Text

Abstract
Background Asthenoteratospermia, one of the most common causes for male infertility, often presents with defective sperm heads and/or flagella. Multiple morphological abnormalities of the sperm flagella (MMAF) is one of the common clinical manifestations of asthenoteratospermia. Variants in several genes including DNAH1, CEP135, CATSPER2 and SUN5 are involved in the genetic pathogenesis of asthenoteratospermia. However, more than half of the asthenoteratospermia cases cannot be explained by the known pathogenic genes.
Methods and results Two asthenoteratospermia-affected men with severe MMAF (absent flagella in >90% spermatozoa) from consanguineous families were subjected to whole-exome sequencing. The first proband had a homozygous missense mutation c.188G>A (p.Arg63Gln) of DZIP1 and the second proband had a homozygous stop-gain mutation c.690T>G (p.Tyr230*). Both of the mutations were neither detected in the human population genome data (1000 Genomes Project, Exome Aggregation Consortium) nor in our own data of a cohort of 875 Han Chinese control populations. DZIP1 encodes a DAZ (a protein deleted in azoospermia) interacting protein, which was associated with centrosomes in mammalian cells. Immunofluorescence staining of the centriolar protein Centrin1 indicated that the spermatozoa of the proband presented with abnormal centrosomes, including no concentrated centriolar dot or more than two centriolar dots. HEK293T cells transfected with two DZIP1-mutated constructs showed reduced DZIP1 level or truncated DZIP1. The Dzip1-knockout mice, generated by the CRSIPR-Cas9, revealed consistent phenotypes of severe MMAF.
Conclusion Our study strongly suggests that homozygous DZIP1 mutations can induce asthenoteratospermia with severe MMAF. The deficiency of DZIP1 induces sperm centrioles dysfunction and causes the absence of flagella.
- asthenoteratospermia
- centrosome
- DZIP1
- flagella
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Infertility prevents millions of couples from natural conception, thus becoming a major healthy concern.1 Asthenoteratospermia is characterised by decreased sperm motility and obvious morphological abnormalities in sperm head, neck or flagella, causing male infertility.2 3 As early as 2003, homozygous partial deletion in CATSPER2 (MIM: 607249) was first described to be associated with infertility phenotype with abnormal sperm motility and morphology in humans.4 Till now, several genes responsible for different types of asthenoteratospermia have been identified. For example, biallelic SUN5 (MIM: 613942) mutations cause severe acephalic spermatozoa.5DNAH1 (MIM: 603332), CFAP family members and some other genes have been reported to induce human multiple morphological abnormalities of the flagella (MMAF).2 6–21 Mutations in AURKC and DYP19L2 account for most cases of macrozoospermia and globozoospermia, respectively.22–26 These findings demonstrated that asthenoteratospermia has strong genetic heterogeneity and diverse phenotypes.
The centrosome consists of two centrioles, pericentriolar material and centriolar satellites, and is involved in numerous functions such as organisation of the mitotic and meiotic spindle.27–31 During flagellum biogenesis, the flagellar axoneme originates from the sperm centrioles located at the basal bodies.32 Moreover, during human fertilisation, the sperm centrosome organises the sperm aster, which is essential to unite the sperm and oocyte pronuclei, and controls the first mitotic divisions after fertilization.33 34 Therefore, it could be speculated that defects in the proteins shared between sperm centrosome and flagella may disturb the flagellar formation, meiosis and first mitotic divisions after fertilisation.
In this study, by using whole-exome sequencing (WES), homozygous mutations of DZIP1 (DAZ interacting zinc finger protein 1, MIM: 608671) were identified in two unrelated Han Chinese men affected with asthenoteratospermia, who did not carry bi-allelic pathogenic mutations in any of those known genes. The mammalian DZIP1 gene encodes a zinc finger and coiled-coil containing protein, which interacts with the DAZ (deleted in azoospermia) protein,35 and is predominantly expressed in testis. Mouse DZIP1 and its zebrafish homologue protein (Iguana) have previously been reported to be associated with centrosomes and ciliogenesis in cells.36–38 Notably, the two DZIP1-mutated probands consistently presented severe MMAF with predominantly high malformation rates of absent flagella (>90%). The absence of DZIP1 and abnormal signals of centrioles were observed in the spermatozoa from both probands. In parallel, we characterised a Dzip1-knockout mouse model, which resembled the severe MMAF phenotypes. Overall, our study strongly suggests that DZIP1 is required for the formation of both sperm flagella and sperm centrioles, and that homozygous DZIP1 mutation can induce asthenoteratospermia with severe MMAF.
Materials and methods
Subjects and clinical investigation
A cohort of 65 unrelated Han Chinese man preliminarily diagnosed with MMAF were recruited from the First Affiliated Hospital of Anhui Medical University and the Affiliated Suzhou Hospital of Nanjing Medical University in China. Some patients in this cohort were previously described.10 16 17 19 39 40 All 65 men suffered from primary infertility for more than 1 year. Two probands in this study were from consanguineous families. Their ages were 27 (A029 IV-1) and 28 (A0033 IV-1), respectively. No obvious symptoms of other ciliopathies (such as primary cilia dyskinesia, polycystic kidney disease or Bardet–Biedl syndrome) were observed in the two probands by careful clinical examinations (online supplementary figure S1). The two probands will be followed up constantly. The karyotype analysis performed in two cases showed normal somatic karyotypes (46; XY) and no large-scale deletions in the human Y chromosome. Informed consents were obtained from each subject.
Supplemental material
Semen analyses were carried out during routine examination of the individuals according to the WHO guideline (the fifth Edition). Sperm morphology was assessed by modified Papanicolaou staining. At least 200 spermatozoa were examined. The percentages of morphologically abnormal spermatozoa were evaluated according to the WHO guidelines.
WES, bioinformatic analysis and Sanger sequencing
WES and bioinformatic analysis were performed according to our previously described protocols.8DZIP1 mutations identified by WES were validated by Sanger sequencing. PCR primers and protocols used for each individual are listed in online supplementary table S1.
Expression vector construction, cell culture and transfection
To construct the wild-type (WT) and two DZIP1-mutated (p.Arg63Gln and p.Tyr23*) expression plasmids, total RNA was extracted from testicular tissues of the control subject with obstructive azoospermia, and was reverse-transcribed to cDNA. The full-length cDNA was amplified, respectively, by full-length PCR and segmental PCR that induced mutations (primer sequences were provided in online supplementary table S2). Then the amplification was inserted into the pEGFP-C1 vector between the restriction sites of BglII and BamHI. HEK293T cells were cultured in DMEM medium supplemented with 10% Fetal bovine serum (Invitrogen) and 1% antibiotics (100 units/mL penicillin and 100 µg/mL streptomycin, Invitrogen) at 37°C and 5% CO2. The empty and recombinant plasmids were transfected into HEK293T cells using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s recommendations.
Western blotting
Transfected cells and half of a mouse testis were homogenised in 200 µL RIPA (Beyotime) using a pellet pestle motor homogeniser and then heated at 100°C for 15 min. Lysates were fractionated by SDS-PAGE on 10% polyacrylamide gels, transferred to polyvinylidene fluoride membranes, and the membranes were blocked in TBST (3% Bovine serum albumin in tris-buffered saline with Tween-20) for 1 hour at room temperature (RT). Anti-DZIP1 antibody (mouse monoclonal, Santa Cruz) was diluted 1:1000 in TBST and incubated with the membranes overnight at 4°C. ECL (Kodak) was used for visualisation. Protein levels were normalised using the reference protein GAPDH.
Transmission electron microscopy
Sample preparation of the human sperm for routine transmission electron microscopy (TEM) has been previously described.17 In this study, more than 1000 sections were observed to investigate the longitudinal sections of sperm and confirm the phenotype of absent axoneme.
Generation of Dzip1-knockout mouse model
Dzip1-knockout mice were generated by Nanjing Biomedical Research Institute of Nanjing University according to our previously published protocol.10 The single-guide RNA (sgRNA) was designed against Dzip1 exon 2. The frameshift mutation in Dzip1 was identified in founder mice and their offspring by Sanger sequencing (the primer information was provided in online supplementary table S3). All experiments involving mice were performed according to the methods approved by the Animal Ethics Committees of each corresponding institution.
Mouse histology
Fresh mouse testis and epididymis were fixed in modified Davidson’s fluid (50% diluted water, 30% formaldehyde, 15% ethanol and 5% glacial acetic acid), respectively, for over 48 hours. After fixed, the tissues were dehydrated in the gradient alcohol (70% ethanol for 24 hours, 80% ethanol for 2 hours, 90% ethanol for 2 hours and 100% ethanol for 1 hour). Then, the tissues were placed in xylene for 1 hour, and finally embedded in paraffin wax and sectioned to ~4 µm.
For haematoxylin and eosin staining, sections deparaffinised in xylene at 65°C overnight. After deparaffinisation, slides were stained with hematoxylin and eosin, dehydrated and mounted.
Immunofluorescence staining
Immunofluorescence experiments were performed using human spermatozoa and mice testes as previously described.10 Briefly, human spermatozoa were coated on the slides and fixed in cold methanol for 5‒10 min. The slides were soaked (3% Bovine serum albumin and 0.1% Triton X-100 in 1× phosphate-buffered saline (PBS)) at RT for 1 hour and then incubated overnight at 4°C with the following primary antibodies: rabbit polyclonal anti-DZIP1 (Abgent, targeting 568–596 amino acids of human DZIP1 protein), rabbit polyclonal anti-IFT88 (Proteintech), rabbit polyclonal anti-IFT140 (Proteintech) and rabbit polyclonal anti-Centrin1 (Proteintech). After that, slides were washed by 1×PBS three times for ten minutes at a time, followed by 1-hour incubation at RT with secondary antibodies (AlexaFluor 647 anti-Rabbit, Yeasen; AlexaFluor 488 anti-Mouse, Invitrogen) and 0.5% 4',6-diamidine-2-phenylindoles (DAPI).
For immunofluorescence staining of mouse testis, after being carefully deparaffinised and rehydrated, the tissue sections were put into boiled 10 mM citrate buffer (pH 6.0) for 10 min and were then treated with 0.1% Triton X-100 in PBS for 30 min. After blocking non-specific binding sites with 10% normal donkey serum (Jackson ImmunoResearch Labs), the slides were incubated with mouse monoclonal anti-DZIP1 (Santa Cruz) at 4°C overnight, and with AlexaFluor 647 anti-Rabbit and 0.5% DAPI for two additional hours at RT. Slides of both sperm and testicular tissues were observed with an LSM800 confocal microscope (Carl Zeiss AG).
Mating test
Fertility was investigated in the male mice (8–12 weeks, n=6) of WT, heterozygous and homozygous Dzip1 mutations. Each male mouse was caged with two WT C57BL/6J females (8–12 weeks), and vaginal plugs were checked every morning. Once a vaginal plug was identified, the male mouse was allowed to rest for 2 days before another female mouse was placed in the cage. The mated female mouse was separated and single caged, and the pregnancy results were recorded. The fertility test lasted for 3 weeks.
Results
Identification of homozygous DZIP1 mutations in men with severe MMAF
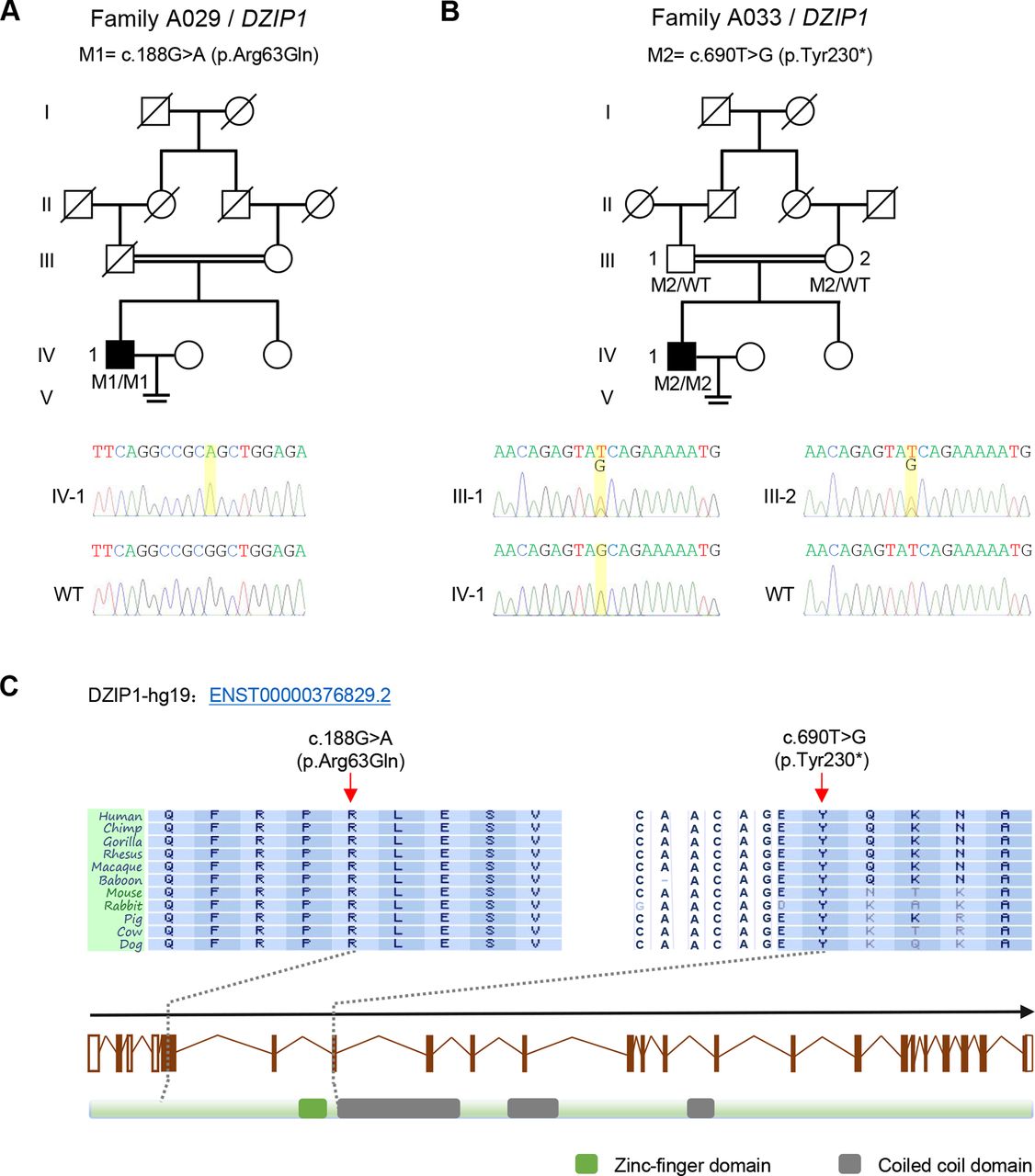
After WES, stringent bioinformatic filtering analysis and Sanger sequencing, we identified homozygous mutations of DZIP1 in two Han Chinese men affected with asthenoteratospermia (figure 1 and online supplementary table S4). The proband IV-1 in the consanguineous family A029 had a homozygous missense mutation c.188G>A (p.Arg63Gln) of DZIP1 (figure 1A). This missense mutation altered a highly conserved amino acid of DZIP1 protein (figure 1C), and was predicted to be potentially deleterious by all three functional prediction tools: SIFT, PolyPhen-2 and MutationTaster. In the other consanguineous family A033, a homozygous stop-gain mutation c.690T>G (p.Tyr230*) of DZIP1 was identified in proband IV-1 and both parents are heterozygous carriers (figure 1B). This mutation was located at the beginning of the first coiled-coil domain of DZIP1 protein, leading to absence of three coiled-coil domains, which are potentially important in protein-protein interactions. As shown in online supplementary table S4, the two DZIP1 mutations were absent from the human population datasets of the 1000 Genomes Project, the Exome Aggregation Consortium and the Genome Aggregation Database. We also investigated a Han Chinese control population consisting of 300 fertile Han Chinese individuals and 668 non-MMAF-affected cases. Notably, both DZIP1 mutations were also absent from the ethnically matched control population (online supplementary table S4). For both probands, no variants with low frequencies in control populations were identified in other genes reported to be associated with cilia, flagella or male infertility. Therefore, DZIP1 appeared to be a new gene involved in MMAF.

Homozygous DZIP1 mutations identified in men with asthenoteratospermia. (A) A missense mutation c.188G>A (p.Arg63Gln) of DZIP1 was identified in the consanguineous family A029. The proband (IV-1) was homozygous for this mutation (M1). (B) a stop-gain mutation c.690T>G (p.Tyr230*) of DZIP1 was identified in the consanguineous family A033. This mutation (M2) was homozygous in the proband (IV-1), and was confirmed to be inherited form his parental heterozygous carries. (C) These two DZIP1 mutations (M1 and M2) are located at the conserved sites close to the N-terminal. Green and grey squares, respectively, stand for zinc-finger domain and coiled-coil domains as described by the UniProt server. Both mutations were verified by Sanger sequencing. The mutation positions are indicated by yellow boxes. Mutations are annotated in accordance to the Human Genome Variation Society’s recommendations. WT, wild type.
In vitro effects of these two homozygous DZIP1 mutations were also investigated in HEK293T cells transfected with WT or DZIP1-mutated constructs. As shown in online supplementary figure S2, the expression level of DZIP1 was obviously reduced for the p.Arg63Gln mutation, and DZIP1 was truncated for the p.Tyr230* mutation.
Asthenoteratospermia with severe MMAF in men with homozygous DZIP1 mutations
The detailed phenotypes in two probands with homozygous DZIP1 mutations were analysed. Semen analysis showed that the sperm concentration was decreased compared with the standard value of WHO (table 1). The spermatozoa of both probands consistently presented as immotile (table 1). Moreover, no spermatozoa with normal morphology could be observed in any of the two DZIP1-mutated men (table 2) under light microscopy. Compared with the spermatozoa from a control sample, various morphological abnormalities of spermatozoa from DZIP1-mutated men were observed, such as short and absent flagella (figure 2). Notably, the major flagellar malformation in DZIP1-mutated men was absent flagella, accounting for more than 90% spermatozoa, which was obviously higher than those of the control fertile men (table 2). In addition, the severe flagellar abnormalities were confirmed by TEM analysis (online supplementary figure S3). Compared with the normal connecting piece and axoneme in the spermatozoon of a control subject, the longitudinal sections of the spermatozoa from the DZIP1-mutated men presented with absent or very short axoneme.

Sperm morphology in DZIP1-mutated men. (A) Normal morphology of a spermatozoon from a healthy control man. (B) to (D) Most spermatozoa of DZIP1-mutated men presented multiple morphological abnormalities, such as very short flagella (B), absent flagella (C) and short flagella (D). scale bar: 5 µm.
Semen characteristics in men with homozygous DZIP1 mutations
Sperm morphology in DZIP1-mutated men
Location of DZIP1 in sperm head and neck, and DZIP1 loss in flagellar and centrosomal malformations
To further investigate the pathogenicity of the identified DZIP1 variants, the localization of DZIP1 and the intraflagellar transport (IFT) proteins (eg, IFT88 and IFT140) in spermatozoa from control and two DZIP1-mutated men were detected by immunofluorescence staining (figure 3). In a spermatozoon from the control individual, the DZIP1 immunostaining was concentrated in sperm head and neck, and the immunostaining of IFT88 and IFT140 was observed in sperm head and tail. Normal axoneme was presented by the staining of acetylated-α-tubulin. However, the spermatozoon from two probands had no organised axoneme and DZIP1 immunostaining. The immunostaining of IFT88 and IFT140 was weak or misplaced in sperm head, and absent in sperm tail.

Immunostaining of DZIP1, IFT88 and IFT140 in human spermatozoa from a healthy control and DZIP1-mutated men. (A) In the fertile control, DZIP1 immunostaining (red) was concentrated in sperm head and sperm neck. However, DZIP1 immunostaining was absent in the spermatozoa from DZIP1-mutated men. (B) IFT88 immunostaining (red) was observed in sperm head and flagellum in the control subject whereas it was dispersive or absent in the spermatozoa from DZIP1-mutated men. (C) IFT140 immunostaining (red) was observed in the middle of sperm head and the flagellum in the control subject, but it was abnormally located in the top of sperm head and sperm neck of the spermatozoa from DZIP1-mutated men. Abnormal axonemes (very short or absent flagella) were showed by abnormal acetylated-α-tubulin staining (A to C, green). All these observations of immunostaining were consistent in the spermatozoa of the two DZIP1-mutated men. scale bar: 10 µm.
As mentioned above, DZIP1 has been reported to be associated with centrosomes. To investigate whether DZIP1 deficiency affects centrosomes, immunofluorescence staining of the centriolar protein Centrin1 was carried out (figure 4). In spermatozoa of a control individual, two angled centriolar dots were observed in sperm neck, and the distal one connected with axoneme. Interestingly, the spermatozoa of proband A033 IV-1 presented the abnormalities including two centriolar dots with abnormal angle, no concentrated dot, or more than two centriolar dots. None of the DZIP1-mutated spermatozoa had well-shaped axonemes. Centriolar dots were counted for one hundred spermatozoa in each individual (figure 4B to D). More than half of spermatozoa had abnormal numbers of centrioles in subject A033 IV-1 with DZIP1 deficiency, which was a significant increase when compared with that in the control.

Immunofluorescence staining of centrin1 showed disorders of centrioles in spermatozoa from the DZIP1-mutated proband A033 IV-1. (A) Spermatozoa from a healthy control man and the proband A033 IV-1 were stained with anti-Centrin1 (green) and anti-acetylated tubulin (red) antibodies. DNA was counterstained with 0.5% DAPI. in the control subject, two angled centriolar dots were observed in sperm neck, and the distal one connected with axoneme. Most spermatozoa of the proband A033 IV-1 presented two centriolar dots with abnormal angle, no concentrated dot, or more than two centriolar dots, and none of them had the well-shaped axoneme. scale bar: 10 µm. (B) to (D) More than 100 spermatozoa from each subject were used for counting Centrin1 dots. In the proband A033 IV-1, more than half of spermatozoa had abnormal numbers of centrioles, and the proportion of spermatozoa with abnormal numbers (n=0 and n>2) of Centrin1 dots was significantly higher than that in the control subject.
Consistent asthenoteratospermia phenotypes in Dzip1-knockout male mice
To investigate the impact of DZIP1 deficiency on spermatogenesis, a frameshift mutation (c.102_132del) was generated in mouse ortholog Dzip1 using the CRSIPR-Cas9 technology (online supplementary figure S4A). This Dzip1 mutation was predicted to cause premature translational termination (p.Ala35Serfs*13) (online supplementary figure S4A), which is closer to DZIP1 N-terminus. Western blotting and immunofluorescence using mouse testes confirmed the absence of DZIP1 in Dzip1-knockout male mice (Dzip1‒/‒) (online supplementary figure S4B, C). The DZIP1 staining was detected in germ cells, especially in elongated spermatids, from the testicular tissue of the WT male mice (online supplementary figure S4C), suggesting that DZIP1 is implicated in spermatogenesis.
Remarkably, all the Dzip1‒/‒ male mice were infertile (figure 5A). Therefore, the reproductive phenotypes of Dzip1-mutated male mice were carefully examined (figure 5). Few spermatozoa from epididymides of the Dzip1‒/‒ male mice were collected (figure 5B), and no motile spermatozoa was observed (figure 5C). Very few epididymal spermatozoa were observed under light microscope after concentration, presenting severe morphological abnormalities mainly with absent flagella, which were consistent with the phenotypes observed by haematoxylin and eosin staining in testis and epididymis (figure 5D). Moreover, no obvious difference of above reproductive phenotypes was observed between the heterozygous mutated (Dzip1+/‒) male mice and the WT male mice (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Severe asthenoteratospermia phenotypes in Dzip1-knockout male mice. (A) The pregnancy rate of heterozygous mutated (Dzip1+/‒) and homozygous mutated (Dzip1‒/‒) male mice. No significant difference in pregnancy rate was observed between the Dzip1+/‒ male mice and the WT male mice. but all the Dzip1‒/‒ male mice were infertile. (B) Sperm counts per epididymis of Dzip1+/‒ and Dzip1‒/‒ male mice. No significant difference in sperm count was observed between WT and Dzip1+/‒ male mice, whereas few spermatozoa from the epididymides of Dzip1‒/‒ male mice were collected. (C) Sperm motility of Dzip1+/‒ and Dzip1‒/‒ male mice. No significant difference in sperm motility was observed between WT and Dzip1+/‒ male mice. But spermatozoa from Dzip1‒/‒ male mice presented no motility. (D) Sperm morphology and histological staining of WT, Dzip1+/‒ and Dzip1‒/‒ male mice under light microscopy. The sperm morphology was normal in WT and Dzip1+/‒ male mice. However, most spermatozoa from Dzip1‒/‒ male mice had severe malformations, such as absent flagella, cytoplasm residual and abnormal heads. In comparison to the WT and Dzip1+/‒ male mice, few spermatozoa with normal flagella were observed in seminiferous tubules and epididymis from Dzip1‒/‒ male mice. Scale bar: 5 µm. abbreviation: NS, no significance; WT, wild type.
Discussion
As mentioned above, we identified homozygous DZIP1 mutations in 2 (3.1%) out of 65 unrelated Han Chinese men affected with asthenoteratospermia. Notably, there are obvious differences in semen parameters and sperm morphology between DZIP1-mutated men and the reported asthenoteratospermia cases in our cohort. The total sperm counts of the two DZIP1-mutated patients were obviously decreased (less than 21 million/ejaculate), while those of the patients with other mutations were reduced in only a portion of cases, or even totally normal. Remarkable reductions in sperm motility (less than 22%) and progress motility (less than 10%) were observed among all the DZIP1-mutated and reported MMAF patients in our cohort. In addition, the extremely high proportion (90%) of absent flagella, which were confirmed through the absence of Tubulin immunostaining and the sperm TEM analysis of longitudinal sections, were identified in the spermatozoa of both DZIP1-mutated men, whereas short flagella were frequently observed in the spermatozoa of FSIP2-mutated (82%) and TTC21A-mutated patients (more than 63%).16 17 Besides, the patients with other gene deficiencies (including CFAP43-mutated, CFAP44-mutated, CFAP69-mutated, CFAP251-mutated, DNAH1-mutated and SPEF2-mutated men) presented multiple malformation of flagella (eg, absent, short and/or coiled flagella) without extreme priority in one specific morphology.10 19 39 40 Furthermore, sperm TEM analyses also revealed the different patterns of ultrastructural abnormalities in sperm among different mutated men. The sperm of DZIP1-mutated men mostly presented with absent or very short axoneme, which leaded to the technical challenge in observing cross-sections of the axoneme. While the majority of the patients with other gene deficiencies in our cohort had observable cross-sections, which presented with relatively minor abnormalities, such as disorganisation or absence of central-pair microtubules, peripheral microtubule doublets and fibrous sheaths.10 16 17 19 39 40 The characteristic of severe flagellar malformation in DZIP1-mutated men, thus, reveals a reasonable speculation that DZIP1 may have different actions from previously known asthenoteratospermia-associated genes and its deficiency can lead to severe MMAF phenotypes.
Previous studies in zebrafish embryos41 42 showed that DZIP1 homologue protein (Iguana) localised to the base of primary and motile cilia and was closely associated with the basal bodies. The absence of Iguana completely inhibited the formation of ciliary pits and consequently the axonemal outgrowth.43 As well, in the mouse Dzip1-mutated embryonic fibroblasts lacking primary cilia, both Cep164 and Ninein appendage proteins failed to localise to ciliary appendages, and IFT components (eg, Ift88 and Ift140) were not recruited to basal bodies.36 Furthermore, in our study, few immunofluorescence signals of tubulin were observed in spermatozoa of DZIP1-mutated men, and immunofluorescence staining signals of IFT88 and IFT140 were also undetected in sperm tails (figure 3). These studies consistently suggest that DZIP1 plays a critical role in the early primary ciliogenesis and/or spermiogenesis, and DZIP1 deficiency can lead to severe arrests of ciliary/flagellar formation. Besides, DZIP1 is also widely expressed in brain, ovary, kidney and other tissues in addition to testis. Although these two DZIP1-mutated men preliminarily presented with primary infertility without obvious phenotypes of other ciliopathies (online supplementary figure S1), the potential and late-onset abnormalities cannot be readily excluded. The follow-up survey and examination should be conducted continuously for long.
In addition, the relationship between DZIP1 and centrosomes has been hinted before. In mammalian cells, DZIP1 acting as a centrosome protein, mediates assembly of the BBSome-DZIP1-PCM1 complex in the centriolar satellites and regulates the centriolar satellite localisation of the BBSome protein during the cell cycle.37 Centriolar satellites are small, microscopically visible granules that cluster around centrosomes as several vehicles for protein trafficking.44 These granules contain numerous proteins, including above-mentioned PCM1 and BBSome proteins, directly involved in centrosome maintenance and ciliogenesis.44 In mammalian cells, silencing of BBS4, which encodes a core member of BBSome, induced PCM1 mislocalisation, concomitant de-anchoring of centrosomal microtubules, arrest in cell division and apoptotic cell death.45 Interestingly, BBS4-depleted cells always contained replicated centrioles,45 which was quite similar to abnormal signals of Centrin1 observed in spermatozoa of the DZIP1-mutated men (figure 4). Furthermore, the concentrated immunostaining spot of DZIP1 was observed in normal sperm neck where sperm centrioles and pericentriolar matrixes locate. Consequently, the failure of flagellar formation and the disorder of centrioles in spermatozoa of DZIP1-mutated individuals were probably due to the centrosomal damage caused by the absence of DZIP1.
It has been recently reported that, in addition to a proximal centriole, there is an atypical flagellum-attached distal centriole existing in the mature spermatozoon of fertile men.46 Two immunostaining spots of Centrin1 signals observed in spermatozoa of the control subject in our study partially confirmed the existence of two centrioles in mature human sperm as well (figure 4). The distal centriole, which functions as the zygote’s second centriole, is capable of recruiting pericentriolar matrixes, forming a daughter centriole, and localising to the spindle pole during mitosis, thus plays essential roles especially during the first mitosis after fertilisation.46 Another recent study as to dual-spindle formation in zygotes also hinted that sperm centrioles could be associated with keeping parental genomes apart in early mammalian embryos through the participation in forming dual spindles.47 Therefore, the abnormality of sperm centrioles caused by deficiency of DZIP1 may have important influence on subsequent fertilisation and cleavage.
Overall, in humans and mice, we found that the DZIP1 deficiency caused by homozygous DZIP1 mutations resulted in male infertility characterised by asthenoteratospermia with the damage of both flagellar formation and sperm centrioles, thus establishing DZIP1 as a gene responsible for asthenoteratospermia with severe MMAF.
Acknowledgments
We would like to thank the families for participating and supporting this study. We also thank the Center of Cryo-electron Microscopy at Zhejiang University for technical support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
ML, WaL, WC and XN contributed equally.
Contributors YC, FZ, CZ and XH designed the study. XH, SY, HW, JW, YG, QT, DT, JiZ, BS, YZ, YX, PZ, ZW and ZZ provided patients’ data and performed clinical assessments. XH, ML, WL, WC, HC,QL, XN, W-YL, JW, JuZ, YG, Y-JC, ChL, CaL, CY and HS conducted the experiments. HZ, XH, ML,WL, W-YL and FZ analysed the data. WL, RL, XH and FZ wrote the manuscript. YC and FZ were responsible for the study supervision. All authors read and approved the final manuscript.
Funding This study was supported by National Natural Science Foundation of China (81971441, 31625015, 81601340 and 31521003),Special Foundation for Development of Science and Technology of Anhui Province (2017070802D150), Natural Science Foundation of Anhui Province (1708085QC59 and 1908085QH313), Shanghai Medical Center of Key Programs for Female Reproductive Diseases (2017ZZ01016), the Shanghai Municipal Science and Technology Major Project (2017SHZDZX01) and Jiangsu Commission of Health (H2018050).
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval This study was approved by the Ethical Committees of Anhui Medical University and the School of Life Sciences at Fudan University.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. All data relevant to the study are available in public.
Author note YC, FZ, CZ and XH jointly direct this study.