Article Text

Abstract
Background Pseudodiastrophic dysplasia (PDD) is a severe skeletal dysplasia associated with prenatal manifestation and early lethality. Clinically, PDD is classified as a ‘dysplasia with multiple joint dislocations’; however, the molecular aetiology of the disorder is currently unknown.
Methods Whole exome sequencing (WES) was performed on three patients from two unrelated families, clinically diagnosed with PDD, in order to identify the underlying genetic cause. The functional effects of the identified variants were characterised using primary cells and human cell-based overexpression assays.
Results WES resulted in the identification of biallelic variants in the established skeletal dysplasia genes, B3GAT3 (family 1) and CANT1 (family 2). Mutations in these genes have previously been reported to cause ‘multiple joint dislocations, short stature, and craniofacial dysmorphism with or without congenital heart defects’ (‘JDSCD’; B3GAT3) and Desbuquois dysplasia 1 (CANT1), disorders in the same nosological group as PDD. Follow-up of the B3GAT3 variants demonstrated significantly reduced B3GAT3/GlcAT-I expression. Downstream in vitro functional analysis revealed abolished biosynthesis of glycosaminoglycan side chains on proteoglycans. Functional evaluation of the CANT1 variant showed impaired nucleotidase activity, which results in inhibition of glycosaminoglycan synthesis through accumulation of uridine diphosphate.
Conclusion For the families described in this study, the PDD phenotype was caused by mutations in the known skeletal dysplasia genes B3GAT3 and CANT1, demonstrating the advantage of genomic analyses in delineating the molecular diagnosis of skeletal dysplasias. This finding expands the phenotypic spectrum of B3GAT3-related and CANT1-related skeletal dysplasias to include PDD and highlights the significant phenotypic overlap of conditions within the proteoglycan biosynthesis pathway.
- molecular genetics
- clinical genetics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Skeletal dysplasias are a group of over 450 different disorders which mainly affect bone and collagen development.1 2 While these disorders are individually rare, combined they affect 1 in 5000 births each year.3 The disease spectrum ranges from reduced bone growth resulting in shorter stature to severe developmental syndromes and perinatal lethality.1 4
One extremely rare skeletal dysplasia, at the severe end of the spectrum, is pseudodiastrophic dysplasia (PDD; MIM: 264180), which is associated with prenatal manifestation and early lethality. It is characterised by short-limbed short stature at birth, facial dysmorphism, and distinctive skeletal abnormalities including short ribs, mild to moderate platyspondyly, broad ilia with flaring, increased acetabular angle, shortened long bones with metaphyseal flaring, elongation of the proximal and middle phalanges with subluxation of the proximal interphalangeal joints, subluxation of the elbow, and talipes equinovarus.5–11
While PDD shows phenotypic similarity to diastrophic dysplasia (MIM: 222600), there are key differences in the radiographic and chondro-osseous features,7 and further differences are also found at the molecular level. Diastrophic dysplasia has been shown to be caused by biallelic loss-of-function mutations in SLC26A2, 12 13 but no likely causative variants have been identified in this gene in previously described cases of PDD.1 14 At present, the genetic basis of PDD remains unknown; however, as the disorder affects both males and females and has been observed to reoccur in families with unaffected parents, an autosomal recessive inheritance is likely.7
Clinically, PDD is classified as part of the ‘dysplasias with multiple joint dislocations’ subgroup, with the majority of these disorders caused by defects in proteoglycan (PG) biosynthesis.1 PGs are expressed on cellular surfaces and form a key component of extracellular matrices.15 16 They consist of a core protein, with covalently bound glycosaminoglycan (GAG) side chains. These polysaccharide side chains are classified based on the assembly of their disaccharide units, such as chondroitin sulfate (CS) and dermatan sulfate (DS). Since the extracellular matrices provide structure and stability for many tissues (ie, bone and cartilage), defects in enzymes involved in PG synthesis have a major impact on the development and assembly of skeletal and dermatological tissues.16 As a group, the genetic disorders that result in enzymatic defects affecting biosynthesis of the PG’s GAG linker region are called ‘linkeropathies’.17 18
Aiming to identify the genetic cause of this extremely rare condition, PDD, we investigated four patients from two unrelated families clinically diagnosed with the disorder. Genomic analyses revealed mutations in two different established skeletal dysplasia genes. Functional validation with both primary cells and human cell-based overexpression assays demonstrated that PDD represents the most severe phenotypic presentation of defects in PG synthesis.
Methods
Study subjects
This study was performed as part of the National Health and Medical Research Council(NHMRC)-funded Genomic Autopsy Study, family study IDs: PED012 (family 1) and PED042 (family 2).
Genetic analyses
Genomic DNA was isolated from whole blood (patients 1B, 2A and 2B, parental samples and unaffected siblings) or cultured chorion cells (patient 1A). DNA samples from patients 1A, 1B and 2B and their unaffected parents were submitted for whole exome sequencing (WES) at the Centre for Cancer Biology Australian Cancer Research Foundation(ACRF) Genomics Facility. Insufficient DNA was available for WES of patient 2A. Exonic sequences were enriched using the IDT xGen (family 1) or Roche NimbleGen SeqCap EZ MedExome (family 2) kit, and libraries were sequenced (100 bp (family 1) or 150 bp (family 2) paired-end reads) on an Illumina HiSeq (family 1) or NextSeq (family 2). Sequencing data were processed through a pipeline based on Picard, and alignment to the Human Genome Build 37 (hg19) was performed using BWA. SNPs and small insertions/deletions (indels) were called using GATK HaplotypeCaller V.3.4. Default filters were applied to variant calls using the GATK Variant Quality Score Recalibration approach. Candidate variants were confirmed and cosegregation assessed by Sanger sequencing of PCR-amplified genomic DNA from affected and unaffected family members.
Cell culture
Primary dermal fibroblasts were isolated from skin biopsies of patient 1A (16 weeks’ gestation) and gender and age-close controls (control 1: 11 weeks’ gestation; control 2: 21 weeks’ gestation). The biopsy was first cultured in Ham’s F10 medium (Gibco, Australia) with 20% fetal bovine serum and 1% Penicillin-Streptomycin (both Sigma-Aldrich, Australia) at 36.6°C, with 5% CO2. Once established, the fibroblasts were cultured in Basal Eagle’s Medium 1X (Gibco) with 10% fetal bovine serum and 2 mM L-glutamine (both Sigma-Aldrich).
Generation of recombinant protein
Wild-type human B3GAT3 expression vector was generated as described previously.19 In brief, a truncated form of wild-type B3GAT3, lacking the first 43 amino acids, was amplified from human placenta cDNA and inserted into a p3XFLAG-CMV8 vector (Sigma, St Louis, Missouri). Wild-type human CANT1 expression vector was constructed as previously described, with slight modifications.20 Briefly, a truncated form of wild-type CANT1, lacking the first 64 amino acids (including the predicted transmembrane domain), was amplified from human placenta cDNA (Clontech, Mountain View, California). The DNA fragment was subcloned into a p3xFLAG-CMV8 vector, resulting in the fusion of CANT1 to the preprotrypsin leader sequence and the 3xFLAG tag sequence at the N-terminus of the vector. Mutant vectors (containing B3GAT3: Arg169Trp and Arg225*; CANT1: Glu215Lys) were constructed by overlapping extension PCR and mutations confirmed by sequence analysis.
B3GAT3 and CANT1 (wild-type and mutant) vectors were then transiently transfected into HEK293T cells using the FuGENE HD DNA Transfection Reagent (Promega). For B3GAT3, 3 days after transfection, cells were collected and lysed with mammalian protein extraction reagent (M-PER, ThermoFisher Scientific) and lysates incubated with anti-FLAG affinity agarose resin (Wako, Osaka, Japan) for 1–4 hours at 4°C. For CANT1, 3 days after transfection, aliquots of the conditioned media were incubated with anti-FLAG M2 agarose resin (Sigma) for 2 hours at 4°C. To examine the expression of recombinant enzymes, sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting (online supplementary figure S1) were performed using either an anti-FLAG antibody (Wako) (B3GAT3) or an anti-FLAG M2 antibody (Sigma) (CANT1).
Supplemental material
Glucuronyltransferase activity assay
The effect of each B3GAT3 variant (Arg169Trp and Arg225*) on the glucuronyltransferase activity of B3GAT3/GlcAT-I was examined as previously described.19 In brief, the resin-bound enzyme from the cell lysate, uridine diphosphate-14C-labelled glucuronic acid (UDP-[14C]GlcA, PerkinElmer) and galactoseβ1-3galactoseβ1-O-methyl (Galβ1-3Galβ1-O-Me, Sigma) were used as the enzyme source, the donor substrate and the acceptor substrate, respectively. The reaction mixture was incubated for 4 hours at 37°C (HEK293T) or 30°C (fibroblasts). Radiolabelled products were separated from UDP-[14C]GlcA using anion-exchange resin, AG 1-X8 (PO4 2− form), as described previously.21 The separated products, [14C]GlcAβ1-3Galβ1-3Galβ1-O-Me, were quantified by a liquid scintillation counter (LSC-7400, Aloka).
Biosynthesis of GAG side chains
The impact of the B3GAT3 variants (Arg169Trp and Arg225*) on the function of B3GAT3/GlcAT-I in the biosynthesis of disaccharide units was investigated using cell-based ELISA (In-Cell ELISA). As described previously,22 CS and DS chains on the cell surface and extracellular matrix were compared between fibroblasts from patient 1A and control subjects. Briefly, fibroblasts were cultured in 96-well plates (5000 cells/well) for 1 day. Cells were washed and treated with chondroitinase ABC (Seikagaku) at 37°C for 30 min, before being fixed and incubated with the primary antibodies, a mixture of anti-CS-stub antibodies (1B5, 2B6 and 3B3) (Cosmo Bio, Tokyo). Cells were then incubated with the secondary antibody, alkaline phosphatase-conjugated anti-mouse IgG, before finally being incubated with the substrate, p-nitrophenyl phosphate, and analysed by measuring the absorbance at 405 nm with an iMark Microplate Absorbance Reader (Bio-Rad).
Nucleotidase activity assay
The effect of the CANT1 variant (Glu215Lys) on the nucleotidase activity of CANT1 towards uridine diphosphate (UDP) was examined, similar to previously described.20 In brief, the assay mixture contained 5 µL of 10-times diluted conditioned media, 50 mM 2-(N-morpholino) ethanesulfonic acid-NaOH (pH 6.5), and 1 mM CaCl2, with 0.2 mM UDP (Promega) as the substrate, in a total volume of 50 µL. The mixture was incubated at 37°C for 10 min, then inactivated by incubation at 95°C for 3 min. To determine the amount of unreacted UDP, the reaction product was mixed with the UDP detection reagent, containing an enzyme to convert UDP to ATP (UDP-Glo Glycosyltransferase Assay Kit, Promega). The resultant ATP was measured using a luciferase/luciferin reaction and the luminescent signals detected using a Victor X4 luminometer (PerkinElmer).
Results
Clinical description
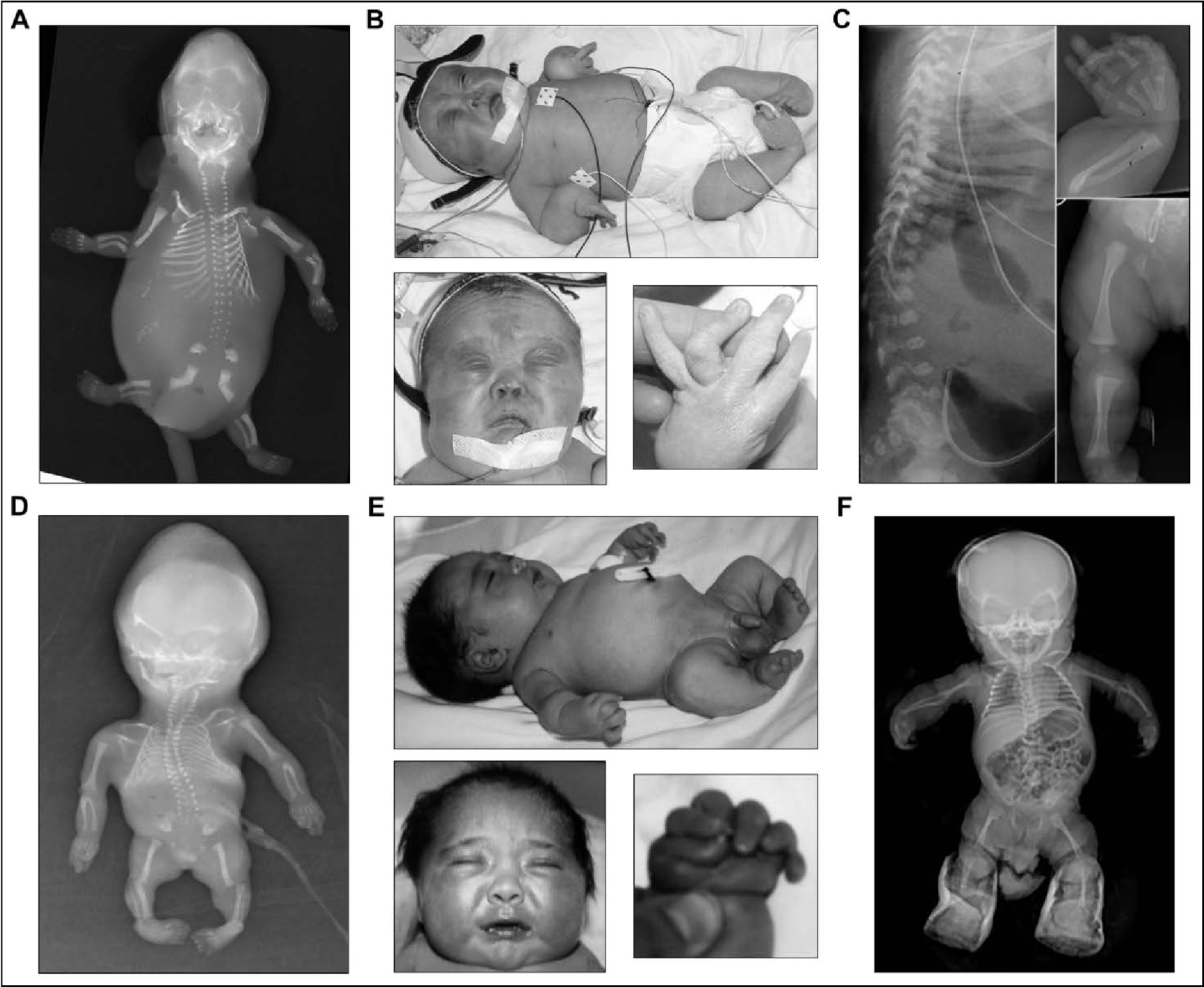
Patients 1A and 1B of family 1 were born to a non-consanguineous couple with three healthy children (two male, one female), described previously11 (figure 1A). Patient 1A was male, stillborn at 16 weeks’ gestation after an uneventful first trimester. Autopsy revealed an oedematous, growth-restricted, dysmorphic fetus with cleft palate, abdominal distension, intestinal malrotation, anorectal dysgenesis, absent external genitalia, and renal system anomalies including enlarged bladder with no outlet, absent left kidney, and right kidney with reduced nephron number and tortuous ureter. The skeletal phenotype included bowed and shortened limbs (especially of the radii, ulnae and femora), short ribs, platyspondyly, small sacrum, reduced ossification of the pubic bones and cervical vertebral segments, and talipes equinovarus (figure 2A).

The pedigrees and the presence of mutations in two unrelated families with pseudodiastrophic dysplasia. (A) B3GAT3 mutations in a non-consanguineous Australian family with two affected siblings. (B) CANT1 mutations in a consanguineous Turkish family with two affected siblings.

Clinical and radiographic phenotype of siblings from two unrelated families with pseudodiastrophic dysplasia. (A,D) Skeletal phenotype of patient 1A (A) and patient 2B (D) at 16 and 18 weeks’ gestation, respectively. Both fetuses appeared hydropic with shortened and bowed long bones, metaphyseal flaring, multiple joint dislocations, flared iliac wings, hypoplastic distal ilia, and talipes equinovarus. (B,E) Clinical phenotype during infancy of patient 1B (B) and patient 2A (E). Both infants displayed relative macrocephaly, short neck, micromelia, short stature, multiple large joint subluxations, interphalangeal dislocation and deviation, and talipes equinovarus. Facial gestalt included round face and full cheeks, prominent eyes, depressed nasal bridge with small anteverted nose, small mouth, and micrognathia. (C,F) Skeletal phenotype during infancy of patient 1B (C) and patient 2A (F). Both infants showed shortened and mildly bowed long bones, with widened metaphyses. Patient 1B also displayed platyspondyly, thoracic kyphosis, and broad and flared ilia. Patient 2A also showed a narrowed thorax, steep acetabular angle and medial metaphyseal beaking of the proximal femora. (A,C) Republished with permission from Yap et al.11 (B) Republished with permission from Spranger et al.40
Patient 1B was female, live-born at 37+5 weeks’ gestation after induction of labour for polyhydramnios. Antenatal ultrasound at 13 weeks’ gestation showed raised nuchal translucency of 4.7 mm, oedema and short femur lengths, which persisted in subsequent scans. Bowing of the long bones was prominent. The postnatal phenotype included generalised micromelia, joint contractures and facial dysmorphism with midface hypoplasia, small upturned nose, micrognathia, small mouth, and cleft palate (figure 2B). Several bilateral anterior polar cataracts were also identified. The distinctive skeletal phenotype included mild platyspondyly, kyphosis, flared ilia, short long bones with metaphyseal flaring, short and broad metacarpals and phalanges, bilateral equinovarus deformity, and multiple joint dislocations (figure 2B,C). These findings were clinically consistent with PDD and allowed retrospective diagnosis of the disorder in patient 1A. Following respiratory distress, patient 1B required invasive ventilation and prolonged intensive care; however, care was withdrawn and the patient was deceased at 9 months of age.
Patients 2A and 2B of family 2 were born to a consanguineous Turkish couple (figure 1B). Patient 2A was male, born at 40 weeks’ gestation after an uneventful pregnancy. Antenatal ultrasonography showed short long bones and increased nuchal thickness. Postnatally, he appeared hydropic with relative macrocephaly, prominent eyes, depressed nasal bridge, small nose, midface hypoplasia, small mouth and a short neck (figure 2E). He had a narrowed thorax and prominent abdomen, short extremities, over-riding and flexion contracture of the fingers, postaxial polydactyly on the right hand, and bilateral talipes equinovarus (figure 2E). Postnatal skeletal survey showed shortened long bones with mild bowing and widened metaphyses, medial metaphyseal beaking of the proximal femora, steep acetabular angle, and multiple joint dislocations (figure 2F). Abdominal ultrasonography was normal. Echocardiography showed ventricular septal defect, secundum atrial septal defect and right-sided arcus aorta. The patient was deceased at 6 months of age due to respiratory insufficiency, secondary to pneumonia.
Patient 2B was also male, terminated at 18 weeks’ gestation after detection of similar manifesting features on ultrasound. Postmortem examination reported hydropic appearance, midface hypoplasia, narrow bell-shaped thorax, short limbs, camptodactyly and equinovarus deformity. Salient skeletal features included shortened long bones, multiple joint dislocations, short metacarpals and malaligned short phalanges (figure 2D). These findings led to a clinical diagnosis of PDD in both affected siblings.
Identification of candidate variants
Proband-parent (quad and trio) WES was employed on families 1 and 2, respectively, resulting in an average coverage of 66x (patient 1A), 70x (patient 1B) and 124x (patient 2B), with 83% (patient 1A), 85% (patient 1B) and 94% (patient 2B) of targeted bases being covered by at least 20 reads (online supplementary table S1).
For family 1, a total of 83 395 (patient 1A) and 82 257 (patient 1B) variants were detected in the WES data. Filtering for rare (<1%), non-synonymous variants resulted in 383 (patient 1A) and 297 (patient 1B) variants. No overlapping homozygous variants were identified. However, overlapping compound heterozygous variants were identified in three genes (SPEG, EID3 and B3GAT3). Of these three genes, only mutations in B3GAT3 are known to cause skeletal dysplasia. B3GAT3 encodes for beta-1,3-glucuronyltransferase 3 (GlcAT-I), and mutations in this gene are known to cause ‘multiple joint dislocations, short stature, and craniofacial dysmorphism with or without congenital heart defects’ (JDSCD (MIM: 245600); formerly Larsen syndrome).19 Segregation analysis by Sanger sequencing confirmed both B3GAT3 variants to be present in compound heterozygosity in the two affected patients. The maternal variant, p.Arg225* (Chr11(GRCh37):g.62384214G>A, NM_012200.3:c.673C>T), and the paternal variant, p.Arg169Trp (Chr11(GRCh37):g.62384572G>A, NM_012200.3 :c.505C>T), were confirmed as heterozygous. Only the paternal variant was present in the two unaffected brothers, with the unaffected sister carrying neither variant (figure 1A).
Both B3GAT3 variants are rare in the population database gnomAD and have never been observed in homozygosity (Arg225*: 4 alleles, 0.001%; Arg169Trp: 3 alleles, 0.001%).23 The Arg225* nonsense variant is predicted to lead to a truncated or absent protein. The variant has been reported once in ClinVar as likely pathogenic, but no functional evidence was provided, and the zygosity and phenotype associated with the variant were not documented (VCV000620480.1). The novel Arg169Trp missense variant is predicted to be deleterious by multiple lines of computational evidence, with Arg169 being highly conserved (97 of 99 species).
For family 2, patient 2B, a total of 94 047 variants were detected in the WES data. Filtering for rare (<1%), non-synonymous, homozygous variants resulted in 24 variants. Only one of these variants affected a gene (CANT1) which has been associated with a skeletal dysplasia phenotype. CANT1 encodes for the enzyme calcium-activated nucleotidase 1, and mutations in CANT1 are associated with Desbuquois dysplasia 1 (DBQD1; MIM: 251 450).24 No compound heterozygous variants were identified in genes associated with other skeletal dysplasias. Segregation analysis by Sanger sequencing showed the CANT1 variant, p.Glu215Lys (Chr17(GRCh37):g.76991292C>T, NM_138793.3:c.643G>A), was also present in homozygosity in patient 2A, confirming segregation with disease. Both parents carried the variant in heterozygosity (figure 1B).
The Glu215Lys missense variant has not been reported before in association with disease, but is rare in the population database gnomAD (1 allele, 0.0004%).23 Glu215 is highly conserved (92 of 92 species) and multiple lines of computational evidence predict the variant to be damaging.
Glucuronyltransferase activity
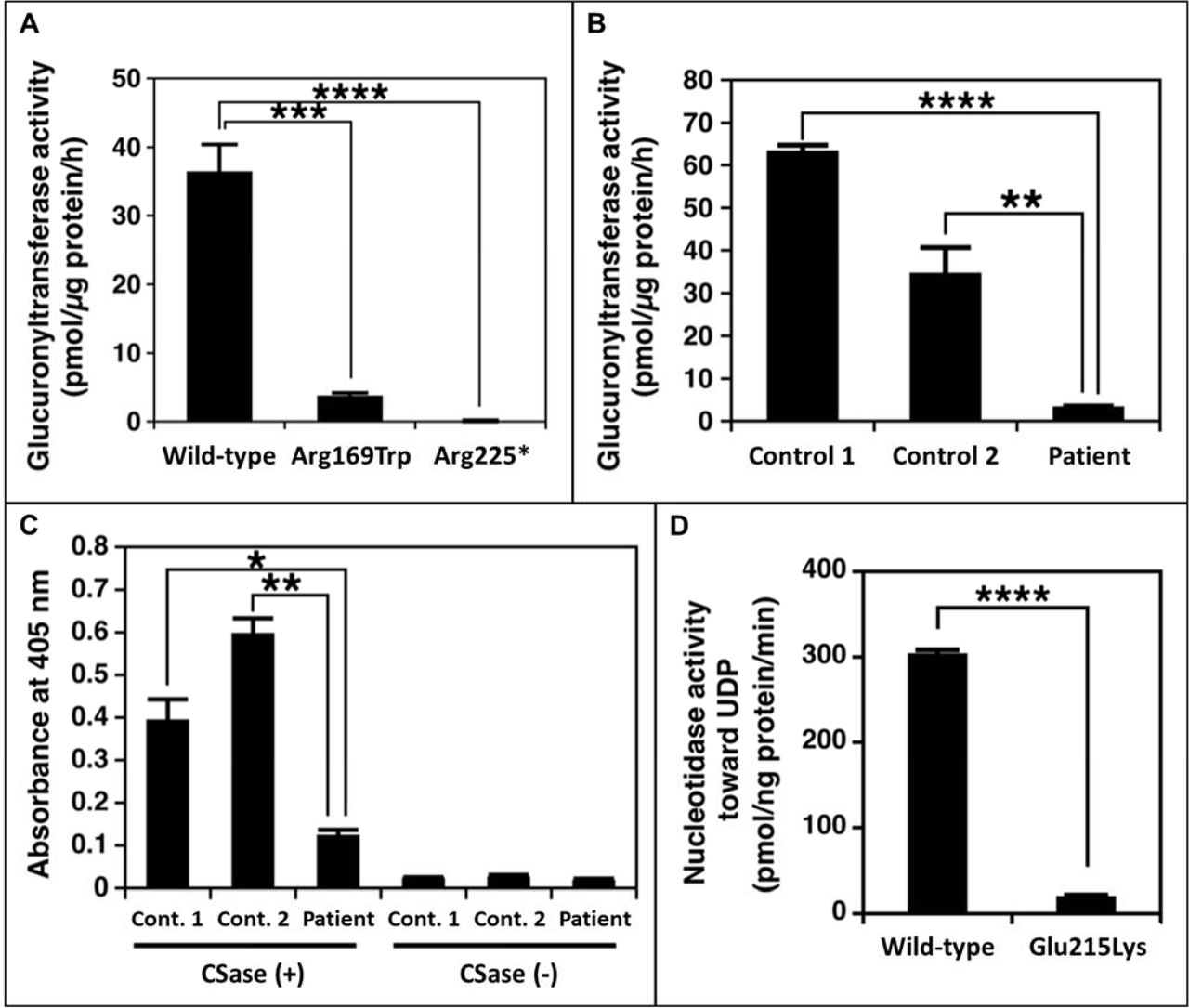
The effect of the B3GAT3 variants (identified in family 1) on glucuronyltransferase activity was assessed using lysates from transiently transfected HEK293T cells and primary fibroblasts. UDP-[14C]GlcA and Galβ1-3Galβ1-O-methyl were used as the donor and acceptor substrates, respectively. This assay revealed significantly (p<0.0002, Student’s t-test) decreased enzymatic activity in recombinant Arg169Trp B3GAT3/GlcAT-I HEK293T cell lysates compared with wild-type cell lysates. Lysates from recombinant Arg225* B3GAT3/GlcAT-I HEK293T cells showed no enzymatic activity (p<0.0001, Student’s t-test) (figure 3A). Similarly, lysates from patient 1A fibroblasts showed significantly (p<0.001, Student’s t-test) reduced glucuronyltransferase activity compared with lysates from control fibroblasts (figure 3B). These results suggest that both variants are severe loss-of-function mutations and confirm that the truncated Arg225* mutant protein is functionally null.
{kind=link}
{kind=link}
{kind=link}
The functional effect of the identified B3GAT3 and CANT1 mutations. (A) Glucuronyltransferase activity of recombinant B3GAT3/GlcAT-I from cell lysates of HEK293T cells. Enzyme activity is significantly decreased for the Arg169Trp mutant protein compared with wild-type protein, and is absent for the Arg225* mutant protein. (B) Glucuronyltransferase activity in fibroblast cell lysates from patient 1A and gender and age-close healthy controls. Enzyme activity is significantly reduced in patient cells compared with controls. (C) In-Cell ELISA result comparing CS/DS chains on the cell surface and extracellular matrix of fibroblasts from patient 1A and gender and age-close healthy controls. CS/DS-stub antibodies are markedly reduced in patient cells compared with controls. (D) Nucleotidase activity of the recombinant CANT1 expressed in HEK293T cells. Enzyme activity is significantly decreased for the Glu215Lys mutant protein compared with wild-type protein. *P<0.01, **P<0.001, ***P<0.0002, ****P<0.0001. CS, chondroitin sulfate; DS, dermatan sulfate; UDP, uridine diphosphate.
Biosynthesis of GAG side chains
The functional impact of the B3GAT3 variants on the biosynthesis of GAG side chains was investigated by analysing CS and DS using a cell-based ELISA (In-Cell ELISA) assay. This experiment revealed markedly (p<0.01, Student’s t-test) reduced CS/DS-stub antibodies in patient 1A fibroblasts compared with controls (figure 3C), indicating a markedly decreased number of CS and DS chains on the cell surface and in the extracellular matrix of patient cells. Taken together with the results of the enzyme activity assay, this indicates that the mutations in B3GAT3 affect glucuronyltransferase activity, leading to a defect in the biosynthesis of GAGs and/or their PGs in the patients.
CANT1 enzymatic activity
The effect of the CANT1 variant (identified in family 2) on nucleotidase activity was evaluated using recombinant protein obtained from conditioned media of transiently transfected HEK293T cells, with UDP used as the substrate. This assay revealed that although the expression levels of wild-type and mutant enzyme in the conditioned media were comparable (online supplementary figure S1B), the enzymatic activity of recombinant mutant CANT1 (Glu215Lys) was significantly (p<0.0001, Student’s t-test) reduced compared with that of wild-type enzyme (figure 3D). This result suggests the variant to be a severe loss-of-function mutation.
Discussion
PDD is known to be an extremely rare disorder at the severe end of the phenotypic spectrum for skeletal dysplasias and is associated with prenatal-onset manifestation and early lethality.7 Considering the lethality, the disorder was long expected to result from a genetic defect in an unidentified disease gene. Aiming to identify this gene and characterise the genetic mutations underlying PDD, we performed exome sequencing on DNA samples from two families (family 1: quad; family 2: trio). Unexpectedly, our genomic analysis revealed mutations in known OMIM genes, B3GAT3 (family 1) and CANT1 (family 2), associated with two different skeletal dysplasias. Consistent with the function of genes causative for other skeletal disorders in the same classification subgroup as PDD, both of these genes are involved in PG synthesis.1
B3GAT3 (GlcAT-I) deficiency is characterised by a defect in glucuronyltransferase activity, resulting in defective biosynthesis of the GAG linker region of PGs.19 Specifically, B3GAT3/GlcAT-I is responsible for the transfer of a GlcA residue from UDP-GlcA to the last of the four saccharides of the linker region.25 Biallelic B3GAT3 mutations are known to cause a skeletal dysplasia phenotype, JDSCD, characterised by shortened and bowed long bones, spine curvatures, and foot deformities. Other common features include facial dysmorphism, joint dislocation, congenital cardiac defects, joint contractures and bone fragility.16 18 19 22 26–31 Lethality before the age of 1 year has also been reported.29 30 The majority of these clinical manifestations were observed in the affected individuals of family 1 and have also been reported in other patients with PDD,5–10 highlighting the extensive phenotypic similarities between PDD and B3GAT3-related disease. Prenatal detection of bowed long bones may be an early indication of a severe B3GAT3-related disorder, like PDD.
The varied clinical spectrum of B3GAT3-associated disease is possibly explained by mutation-specific levels of residual enzyme activity.30 We therefore performed functional assays to evaluate the downstream effect of the identified variants in B3GAT3. For both variants (Arg169Trp and Arg225*), cell lysates from recombinant HEK293T cells showed a near-complete or complete loss of enzyme activity in vitro, respectively (figure 3A). Similarly, the patient fibroblasts showed severely diminished glucuronyltransferase activity and a reduction of CS and DS chains (figure 3B,C). While these experiments provide the required functional evidence to confirm that the B3GAT3 mutations are the cause of skeletal dysplasia in family 1, simultaneous testing of patient fibroblasts from patients with PDD and JDSCD will be required to elucidate genotype–phenotype correlations. However, due to early lethality of PDD and the rarity of both disorders, this might not be feasible.
The CANT1 nucleotidase is known to hydrolyse UDP, a product of glycosyltransferase reactions.20 Functional defects in CANT1 result in impaired removal of UDP, leading to UDP accumulation and inhibition of glycosyltransferase activities, thereby limiting GAG synthesis.32 Biallelic mutations in CANT1 are associated with DBQD1, which is characterised by prenatal-onset short stature, multiple joint dislocations, hand anomalies and advanced carpal ossifications. Additional features can include facial dysmorphism, congenital cardiac defects and talipes equinovarus,24 32–38 again overlapping closely with many of the features observed in patients with PDD.5–10 Interestingly, several of the earlier described CANT1 patients also died perinatally or during early infancy.24 32 Following the identification of CANT1 as the molecular basis of the PDD phenotype in family 2, the subtle presence of monkey-wrench femoral neck in patient 2A was retrospectively considered consistent with CANT1-related disease.
CANT1 mutations have also been identified as causative of a relatively milder skeletal disorder, multiple epiphysial dysplasia 7 (MED7; MIM 617719), characterised by short stature and early-onset osteoarthrosis.39 Functional assessment of these MED7-linked mutations, in parallel with DBQD1 and PDD mutations, will be important for understanding the mechanistic basis of the described allelic heterogeneity. Similar to an earlier study, we assessed the functional effect of the variant we identified in CANT1 by measuring its activity towards UDP.34 The recombinant mutant CANT1 (Glu215Lys) expressed in HEK293T cells showed defective nucleotidase activity, with close to complete loss of function (figure 3D).
In conclusion, we describe four patients from two families with a clinical diagnosis of PDD and mutations in known genes involved in skeletal dysplasias (B3GAT3 and CANT1). Functional assays confirmed causality of the mutations and support previously published functional data which suggest that a complete loss of function is responsible for the severe, lethal phenotype observed in our patients. We propose that PDD is likely not a separate genetic disorder, but rather may be considered the most severe phenotypic manifestation of skeletal dysplasia arising from defects in PG biosynthesis. This finding highlights the clinical challenge in differentiating PDD from other skeletal dysplasias, such as B3GAT3-associated and CANT1-associated disorders, due to significant phenotypic overlaps. A genomics-first approach, like exome sequencing, may therefore be a valuable tool in delineating the molecular defects underlying skeletal dysplasias and will aid in nosology and characterising the subtle phenotypic heterogeneity within this group of disorders.
Acknowledgments
We would like to thank the families for their participation in this study. We also thank Wendy Waters and Karin Kassahn of the Genetics and Molecular Pathology Department, SA Pathology, Adelaide, South Australia, Australia and Ruriko Netsu and Tomoyo Tada of the Department of Pathobiochemistry, Meijo University, Nagoya, Japan for their technical assistance. Thanks also to all the staff of the Genetics and Molecular Pathology Research Laboratory and of the Centre for Cancer Biology ACRF Genomics Facility.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @AliciaBByrne, @peer_arts
ABB and SM contributed equally.
Contributors ABB, SM, PA, PY and RS drafted the manuscript. ABB, MB and SLK-S coordinated the study. JF and AWS processed the WES data, and ABB performed the data analysis. SM, KS and SY performed the functional investigations. ABB, SM, PA, PY, KS, SY, RS and HSS contributed to interpretation and discussion of results. LM performed the surgical pathology investigations. JEL, HM-A, GN, PY and RS provided clinical care. HSS and CPB conceived and supervised the study. All authors read and approved the manuscript.
Funding This research was supported by the NHMRC (APP1123341) and the Australian Genomic Health Alliance NHMRC Targeted Call for Research into Preparing Australia for the Genomics Revolution in Healthcare (GNT1113531) to HSS and CPB; the Australian Cancer Research Foundation to HSS; Grant-in-Aid for the Research Center for Pathogenesis of Intractable Diseases from the Research Institute of Meijo University to SM and SY; and Grant-in-Aid for Scientific Research (C) 19K07054 from the Japan Society for the Promotion of Science to SM. Additional support was provided by Cancer Council SA's Beat Cancer Project on behalf of its donors and the State Government of South Australia through the Department of Health, and NHMRC Fellowship (APP1023059) to HSS; the Australian Government Research Training Program Scholarship and the Australian Genomics Health Alliance PhD Award and NHMRC (GNT1113531) to ABB; the Takeda Science Foundation to SM; and The Hospital Research Foundation Fellowship to PA.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval This study was conducted in accordance with the Declaration of Helsinki, and was approved by the Human Ethics Committee of the Women’s and Children’s Health Network, Adelaide, South Australia, Australia (HREC/15/WCHN/35) and by the local ethics committees of Meijo University, Nagoya, Japan. Informed consent for genomic analysis, further research and publication of photographs was obtained from parents under institutionally approved consent protocols.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Sequence data has been deposited at the European Genome-phenome Archive, hosted by the European Bioinformatics Institute. All unique materials and datasets generated and/or analysed during the current study are available from the corresponding author upon reasonable request.