Article Text

Abstract
Background Familial hemiplegic migraine (FHM) is a rare form of migraine with aura that often has an autosomal dominant mode of inheritance. Rare mutations in the CACNA1A, ATP1A2 and SCN1A genes can all cause FHM revealing genetic heterogeneity in the disorder. Furthermore, only a small subset of the affected individuals has a causal mutation. We set out to investigate what differentiates patients with FHM with no mutation in any known FHM gene from patients with common types of migraine in both familial and sporadic cases.
Methods 2558 male and female participants from a migraine cohort from the Danish Headache Center were included. 112 had FHM; 743 had familial migraine; and 1703 had sporadic migraine. Using a linear regression model, we analysed for over-representation of rare functional variants in FHM versus familial migraine and sporadic migraine. Post hoc analyses included pathway analysis and testing for tissue specificity.
Results We found that patients with FHM have significantly more rare frameshift indels compared with patients with familial migraine and sporadic migraine. Pathway analysis revealed that the ‘ligand-gated ion channel activity’ and ‘G protein-coupled receptor downstream signalling’ pathways were significantly associated with mutated genes. We moreover found that the mutated genes showed tissue specificity towards nervous tissue and muscle tissue.
Conclusion We show that patients with FHM compared with patients with common types of migraine suffer from a higher load of rare frameshift indels in genes associated with synaptic signalling in the central nervous system and possibly in muscle tissue contributing to vascular dysfunction.
- headache (including migraine)
- neurology
- clinical genetics
- genetics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Familial hemiplegic migraine (FHM) is a rare form of migraine with aura that often has an autosomal dominant mode of inheritance.1 The disorder is characterised by migraine with aura that involves reversible motor weakness/hemiparesis, together with at least one affected first-degree or second-degree relative.2 Rare mutations in the CACNA1A, ATP1A2 and SCN1A genes can all cause FHM, revealing genetic heterogeneity in the disorder.3–5 Furthermore, only a small subset of the affected individuals have a causal mutation.6–8 A Danish population-based survey identified mutations in the three FHM genes in 14% of affected FHM individuals, and a Finnish study identified mutations in only 9% of 45 families with FHM.9 10
FHM is rare with an estimated prevalence of 0.003%, but the common types of migraine are complex disorders with a prevalence of at least 15%.9 11–13 Migraine consists of two major types: migraine without aura (MO) and migraine with typical aura (MA). MO is a clinical syndrome characterised by a unilateral and severe headache with associated symptoms like nausea, vomiting, phonophobia and photophobia.2 MA is primarily characterised by transient focal neurological symptoms, known as aura, as well as headache.2
The common types of migraine (MO and MA) have a polygenic nature where genetic variations account for about 50% of migraine susceptibility.11 14–22 Genome-wide association studies have identified common genetic variants associated with MO and MA.22 Polygenic risk scores of migraine have shown that the common variants from genome-wide association studies explain 5.5% of the phenotypic variance in families with MA, 3.5% in families with MO and 8.2% in families with FHM.10 Moreover, research reports the absence of other monogenic mutations in patients with FHM with no mutation in any known FHM gene.8
We hypothesise that multiple functional variants, that is, burden, increase the risk of FHM and that such variants are accumulated in FHM. We therefore investigated what differentiates patients with FHM with no mutation in any known FHM gene from patients with common types of migraine in both familial and sporadic cases. As the known FHM mutations are all rare functional mutations, we focused on rare functional genetic alterations (frameshift indels, non-frameshift indels, stopcodon indels and non-synonymous Single Nucleotide Polymorphism (SNP)) in FHM compared with common types of migraine. We performed whole-genome sequencing (WGS) in 2558 patients with migraine including 112 patients with FHM from 42 families with no rare variants in any of the known FHM genes, 743 familial cases of common types of migraine from 196 families and 1703 unrelated cases of common types of migraine.
Investigating rare functional variants, we detected genes related to synaptic signalling in the nervous system and in muscle tissue in which a higher mutation burden distinguished FHM from common types of migraine.
Methods
Study cohorts
The study population consisted of 2558 male and female participants from a migraine cohort from the Danish Headache Center (DHC), Rigshospitalet Glostrup (online supplementary tables 1 and 2). Recruitment of participants began in 1999 and is still ongoing.11 23–25 Participants were excluded if they had secondary headache related to an underlying brain illness, systemic illness, trauma or injuries, if they had other than Danish ethnicity or if they were not able to participate in the semistructured interview.
Supplemental material
Supplemental material
FHM cohort
We included 112 participants with FHM from 42 families in the analysis. The participants were systematically recruited from the Danish population, as described in detail elsewhere.11 FHM was defined according to the criteria of the International Headache Society, as patients with migraine with aura that included motor weakness and with at least one first-degree relative who also had attacks of migraine with some degree of motor weakness.26 The diagnosis was based on a semistructured validated telephone interview and a neurological examination both performed by a neurological resident.11
Familial migraine cohort
We included 743 participants from 196 families with Mendelian-like segregation of migraine. Of the 743 participants, 489 fulfilled the diagnostic criteria for MA or probable migraine with typical aura (pMA). A total of 253 individuals were diagnosed with MO or probable MO, none of whom fulfilled the diagnostic criteria for MA or pMA. Initially, a proband from each family was recruited from the Danish population or directly through the DHC and, subsequently, family members were recruited to enable segregation analysis.23–25 27 Each participant’s diagnosis was based on The International Classification of Headache Disorder 3rd edition (ICHD-3) criteria according to a validated semistructured interview performed by a trained physician or a senior medical student trained in headache diagnostics.27–29
Sporadic migraine cohort
The cohort of migraine participants without any first-degree relatives affected by migraine, sporadic migraine, consisted of 1703 participants. A total of 752 participants were diagnosed with MA or pMA. A total of 954 individuals were diagnosed with MO or probable MO, of which none fulfilled the diagnostic criteria for MA or pMA. All were recruited from the DHC and diagnosed using the same procedure as described for familial migraine.
Peripheral blood samples for genomic DNA extraction were collected from all participants after written informed consent.
Sequencing and annotation of rare functional variants
Genomic DNA extraction from whole blood and WGS were performed in collaboration with deCODE genetics as described in detail elsewhere.30
Annovar v.2018apr16 was used to annotate variants. Rare variants were defined as variants with a minor allele frequency (MAF) of <5% using gnomAD V.2 as reference. We defined functional variants as stop codon insertions/deletions, frameshift insertions/deletions, non-frameshift insertions/deletions and non-synonymous SNPs. The mean number of rare functional variants, the mean MAF of rare functional variants and the mean number of variants per patient for each patient cohort are listed in online supplementary table 5.
Supplemental material
Segregating rare functional variants in known FHM genes
We used the Pedigree Variant, Analysis, and Search Tool V.231 to confirm that no rare variants were found in the known FHM genes CACNA1A, ATP1A2 and in SCN1A using 10 000 permutations.
Statistical analysis
To identify significant differences in the number of rare functional variants, we fitted a generalised linear mixed model using the Markov Chain Monte Carlo technique. We adjusted for the total number of rare functional variants or all variants, phenotype, gender and kinship using the Mixed modeling with genetic relationship matrices (MCMCgrm) function in the GAP V.1.1–22 R package. We adjusted for kinship using a kinship matrix including all our patients from all three cohorts. The kinship matrix was calculated using the Kinship V.1.1.3 R package with pedigrees as input data. The difference between patient groups was calculated as the difference in the area under the curve (AUC) between models with and without numbers of functional variants. The pROC V.1.14.0 R package was used for AUC analysis. P values were corrected using Bonferroni correction, adjusting for the number of tests (n=3 or 4).
A t-test was used to detect the group of genes with a significantly higher mean frameshift indels per gene in FHM-specific genes compared with FHM genes overlapping with either familial migraine, sporadic migraine or both. A one-sided Fisher’s test was used to detect genes with significantly higher occurrences of frameshift indels in FHM compared with both groups of patients with migraine. P values were corrected using Bonferroni correction, adjusting for the number of genes (n=762, 718 or 646).
Pathway analysis
Panther pathway analysis V.14.1 (http://www.pantherdb.org/tools) was used to detect significantly over-represented pathways. The Reactome Pathway and the PANTHER GO-Slim Molecular Function from the 2018_04 ReferenceProteome dataset were used. P values were corrected using Bonferroni correction.
Tissue-specificity analysis
Tissue-specificity analysis for gene sets was conducted using the TissueEnrich R package.32 We selected 23 tissues from the GTEx dataset included and analysed both for tissue enrichment and tissue enhancement. Twenty-eight, 92 and 73 genes were analysed for tissue specificity against background genes (genes expressed higher than 1 Transcripts Per Kilobase Million (TPM) or Fragments Per Kilobase Million (FPKM)). P values were calculated using a hypergeometric test32 and corrected using Bonferroni correction, adjusting for the number of tissues (n=23).
Results
Distribution of functional variants
We analysed the distribution of rare functional variants in patients with FHM compared with patients with familial migraine and patients with sporadic migraine. We analysed patients with FHM from families with no fully segregating rare functional variants in CACNA1A, ATP2A1 or SCN1A genes. Identification of these families were performed using linkage analysis.
Using linear regression, we found that patients with FHM had significantly more rare functional variants compared with both familial migraine and sporadic migraine (figure 1A–D and table 1). The difference between FHM and familial migraine was, however, smaller compared with the difference between FHM and sporadic migraine.

(A) Boxplot of the percentile distribution of rare functional variants in FHM, Fam. migraine and Spo. migraine. (B–D) MCMCgrm AUC analysis of rare functional variants in FHM versus Fam. migraine, FHM versus Spo. migraine, and Fam. migraine versus Spo. migraine. (E,F) MCMCgrm AUC analysis of rare functional variant subtypes: frameshift indels, non-frameshift indels, stop codon indels and non-synonymous. SNPs, in FHM versus Fam. migraine and FHM versus Spo. migraine. AUC, area under the curve; Fam., familial; FHM, familial hemiplegic migraine; Spo., sporadic; SNP, Single Nucleotide Polymorphism
Patient groups and rare variant groups
We found that familial migraine had significantly more rare functional variants compared with sporadic migraine. Secondary downstream analysis of FHM compared with familial migraine and sporadic migraine is therefore conducted separately.
We grouped the rare functional variants into four groups: frameshift indels, non-frameshift indels, stop codon indels and non-synonymous SNPs, and analysed whether the difference was caused by one or more groups of rare functional variants. We found that the patients with FHM had significantly more rare frameshift indels compared with both familial migraine and sporadic migraine (figure 1E,F, table 1 and online supplementary figure 1). In contrast, we found significantly less rare non-synonymous SNPs in FHM compared with both familial migraine and sporadic migraine. We found no difference in rare non-frameshift indels nor in rare stop codon indels.
There was no difference within the four groups of functional variants between familial MA and familial MO nor between sporadic MA and sporadic MO (online supplementary figures 2, 3 and supplementary table 3).
Identifying causal genes
Focusing on the rare frameshift indels in FHM, we found that 86.4% (n=762) of the FHM genes were overlapping between FHM and familial migraine, and the remaining 13.6% (n=120) genes were specific to FHM (figure 2A). Comparing FHM with sporadic migraine, we found that 81.4% (n=718) of the FHM genes were overlapping and 18.6% (n=164) of the genes were specific to FHM (figure 2C). Comparing FHM with both familial migraine and sporadic migraine, we found that 73.2% (n=646) of the FHM genes were overlapping in all three cohorts, and 5.4% (n=48) were specific to FHM (online supplementary figure 5A).
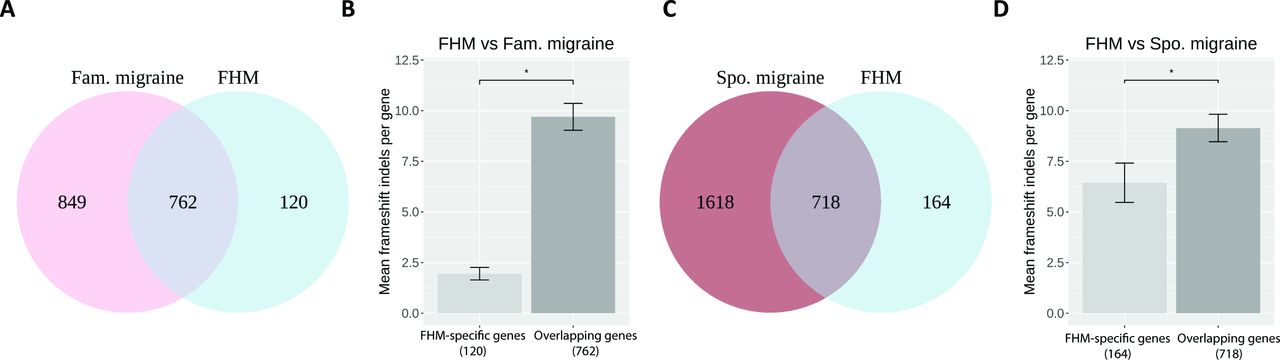
(A) Venn plot showing the number of genes with frameshift indels in FHM, Fam. migraine and the number of overlapping genes. (B) Bar plot with mean frameshift indels per gene in patients with FHM in the 120 FHM-specific genes and in the 762 FHM–Fam. migraine overlapping genes. The asterisk shows a significance level of <0.05. (C) Venn plot showing the number of genes with numbers of frameshift indels in FHM, Spo. migraine and the number of overlapping genes. (D) Bar plot with mean frameshift indels per gene in patients with FHM in the 164 FHM-specific genes and in the 718 FHM–Spo. migraine overlapping genes. The asterisk shows a significance level of <0.05. Fam., familial; FHM, familial hemiplegic migraine; Spo., sporadic.
To pinpoint whether the higher number of frameshift indels in FHM compared with familial migraine and sporadic migraine was driven by the overlapping genes or the FHM-specific genes, we calculated the mean frameshift indels per gene in the respective groups. We found that the contribution was mainly driven by the overlapping genes between FHM and familial migraine having significantly more frameshift indels compared with the FHM-specific genes (p=1.18e-24) (figure 2B). Similar results, though less significant, were obtained for the overlapping genes between FHM and sporadic migraine, which had more mutations compared with the FHM-specific genes (p=0.023) (figure 2D). Similar results were obtained when comparing FHM with both familial migraine and sporadic migraine (p=2.18e-28) (online supplementary figure 5B).
Within the overlapping genes, there were 28 genes having significantly more frameshift indels in FHM compared with familial migraine (padj<0.05); 92 genes showed significantly more frameshift indels in FHM compared with sporadic migraine (padj<0.05) (online supplementary figure 4 and supplementary table 4); and 73 genes showed significantly more frameshift indels in FHM compared to both sporadic migraine and familial migraine (padj<0.05) (online supplementary figure 5C and supplementary table 4).
Supplemental material
Biological relevance of causal genes
To investigate the putative biological mechanisms of the 28, 92 and 73 overlapping genes enriched with rare frameshift indels in patients with FHM compared with patients with familial migraine and sporadic migraine, respectively, we performed pathway analyses and tissue-enrichment analyses.
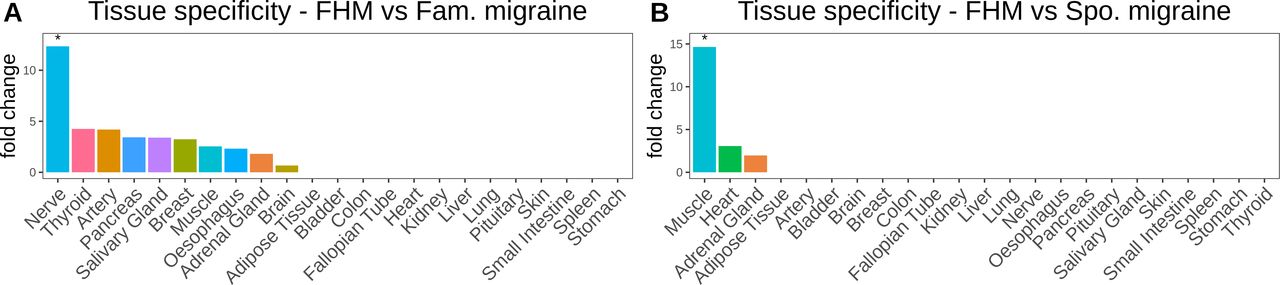
In the 28 genes, we found that the ‘ligand-gated ion channel activity’ pathway was significantly over-represented (padj=0.013) (table 2). Moreover, we found that the 28 genes showed tissue specificity towards the nervous system (padj=0.041) (figure 3A).

(A) Bar plot of gene fold change enrichment of the 28 genes, which had significantly more frameshift indels in FHM compared with Fam. migraine in 23 human tissues from the GTEx RNA-seq database. (B) Bar plot of gene fold change enrichment of the 92 genes, which had significantly more frameshift indels in FHM compared with Spo. migraine in 23 human tissues from the GTEx RNA-seq database. All asterisks display the significance level (padj<0.05). Fam., familial; FHM, familial hemiplegic migraine; Spo., sporadic.
Results from panther pathway analysis
In the 92 genes, we found that the ‘GPCR downstream signalling’ pathway was significantly over-represented (padj=0.027) (table 2) and that the 92 genes showed tissue-specificity towards muscle tissue (padj=7.82e-05) (figure 3B).
We found no pathways significantly associated with the 73 genes. Neither did the genes show any significant tissue specificity. However, we did observe a fold change above 3 and 5 for nerve tissue and muscle tissue, respectively (online supplementary figure 5D).
Discussion
We set out to investigate what differentiates FHM with no known mutation in the CACNA1A, ATP1A2 and SCN1A genes from common types of migraine. We analysed rare functional variants in FHM and compared them with familial migraine and sporadic migraine. The results showed that patients with FHM and no mutation in the known FHM genes have a higher burden of rare frameshift indels compared with common types of migraine. Focusing on rare frameshift indels, we found that the majority of genes with frameshift indels were shared between FHM, familial migraine and sporadic migraine. Notably, the shared genes were significantly enriched with rare frameshift indels compared with genes only present in FHM. It was then possible to conclude that most of the differences between FHM and common types of migraine originate from the shared genes and not from the FHM-specific genes. There were 28 genes with significantly more rare frameshift indels in FHM compared with familial migraine and 92 genes with significantly more rare frameshift indels in FHM compared with sporadic migraine. Pathway analysis revealed an over-representation of the ligand-gated ion channel activity pathway among genes shared between familial migraine and FHM, supported by tissue specificity towards nerves. For FHM and sporadic migraine, the GPCR downstream signalling pathway was enriched with a tissue specificity towards muscle tissue.
Our results suggest that a higher burden of rare frameshift indels is present in FHM compared with common types of migraine, familial as well as sporadic. Frameshift indels shift the translation of the encoded protein. This can result in a protein with an alternative tail, which can be either shorter or longer compared with the wild-type protein. The functional effect is a protein with altered molecular biological function. In rare cases, such proteins can, in an autosomal-dominant way, induce severe phenotypes, including neurological diseases.33–35 Frameshift indels can also act in a recessive way, where an accumulation of frameshift indels is necessary to induce a certain phenotype.36 Our results indicate that FHM in patients without a mutation in CACNA1A, ATP1A2 and SCN1A is possibly caused by an accumulation of rare frameshift indels in multiple genes. This would mean that FHM is not always a dominantly inherited monogenic disease but is caused by the interaction of mutations in multiple genes with moderate effects. This is supported by a family study where patients with FHM and migraine shared the same mutations.37 In this family, FHM was caused by compound heterozygosity, whereas the common forms of migraine were caused by a single mutation in the same gene. However, we cannot exclude that mutations in yet unknown FHM-specific genes contribute to the disease.
Patients with FHM with a mutation in CACNA1A, ATP1A2 and SCN1A often have a more severe phenotype that can include earlier disease onset, more severe hemiparesis, progressive ataxia, mental retardation, cerebrospinal fluid pleocytosis during attacks and brain oedema.8 On the contrary, patients with FHM without a mutation in the known FHM genes have a phenotype more akin to migraine with aura.8 37 As we see a higher burden of frameshift indels in patients with FHM in the genes shared with common types of migraine, this could indicate that FHMs with and without mutations in CACNA1A, ATP1A2 and SCN1A are two different diseases with different clinical characteristics, prognosis and heritability. In our regression analysis, we observed that the difference between FHM and familial migraine was smaller, compared with the difference between FHM and sporadic migraine. Moreover, the percentage of FHM genes with frameshift indels overlapped with a higher percentage to familial migraine compared with sporadic migraine. This suggests that FHM is somewhat more genetically similar to familial migraine compared with sporadic migraine. Taken together, our data fit with previous findings and add to the notion that FHM without a mutation in the known FHM genes might be more closely related to common types of migraine. We found genes shared between FHM and migraine, which had significantly more rare frameshift indels. Contracting a greater burden of these mutations likely contributes to the risk of having FHM compared with the risk of having the milder common migraine phenotype.
It has been suggested that FHM is a genetically heterogeneous channelopathy disorder. CACNA1A and SCN1A encodes voltage-gated ion channels, and ATP1A2 encodes an ion transporter. FHM might thus be caused by faulty synaptic signalling.1 CACNA1A and SCN1A are mainly expressed in Gamma aminobutyric acid (GABA) expressing neurons in the central nervous system (CNS), whereas ATP1A2 is expressed both in CNS and in heart and muscle.38 We found that overlapping genes between FHM and familial migraine with significantly more rare frameshift indels in FHM are associated with the ligand-gated ion channel pathway and that the genes show tissue specificity towards nerves (figure 4A). Ligand-gated ion channel is a broad description that covers multiple classes of ion channels, including GABA receptors. They are expressed throughout the CNS and are located in both the presynaptic terminal and the postsynaptic terminal and contributes significantly to the synaptic signalling. Our data therefore fit the current understanding of the underlying neuronal mechanisms of FHM.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed model of affected signalling and tissues in FHM. We found that following tissues and pathways are affected: (A) Nerve junction with release of neurotransmitters, which act as ligands on the LGICs and subsequently leads to downstream signalling in the postsynapse. (B) Junction between postsynapse and smooth muscle cell. GPCRs are expressed in both cell types and can lead to downstream signalling in both cell types as well. FHM, familial hemiplegic migraine; GPCR, G protein-coupled receptor; LGIC, ligand-gated ion channel.
We furthermore linked the GPCR downstream signalling pathway to the overlapping genes between FHM and sporadic migraine with significantly more rare frameshift indels in FHM. The genes showed tissue specificity towards muscle. GPCRs are a different class of receptors compared with voltage-gated ion channels and ligand-gated ion channels. They are expressed both presynaptically and postsynaptically in the nervous system and in smooth muscle tissue and can regulate synaptic plasticity and signalling (figure 4B). This might be linked to the vascular dysfunction in migraine with constriction of cerebral arteries and the increased risk of cardiovascular events, including myocardial infarction and ischaemic stroke seen in patients with migraine.39 40 From our data, it might be relevant to consider that both neuronal CNS dysfunction driven by ion channel deficits and vascular dysfunction play a role in the pathogenesis of FHM.
In summary, we present novel genetic findings bridging the unresolved genetic gap between FHM without a monogenic mutation and common types of migraine. We show that patients with FHM compared with patients with common types of migraine suffer from a higher load of rare frameshift indels in genes associated with synaptic signalling in the CNS and possibly in muscle tissue contributing to vascular dysfunction. Finally, we suggest that the FHM phenotype in certain cases can be caused by the interaction of mutations in multiple genes with moderate effect instead of one mutation in one gene.
Acknowledgments
We thank Kari Stefansson and Hreinn Stefansson from deCODE Genetics, Iceland, Reykjavík, for genetic analysis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors AR: designed and performed the experiments and wrote the first draft. IAO: designed the experiments and wrote the first draft. MAC: collected data material and provided crucial information. JO: provided clinical information and supervision and raised funding. TFH: designed the experiments, provided supervision, and wrote and edited manuscript.
Funding This project was financed by a grant from Candy's Foundation 'CEHEAD' (JO).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study was approved by the Danish Scientific Ethical Committee (H-2-2010-12) and the Danish Data Protection Agency (i-suite number 01080 and ID number GLO-2010–10).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request as aggregated and anonymized data. Individual data necessitate MTA aggrement.