Article Text

Abstract
Background Since 1994, over 50 families affected by the episodic ataxia type 1 disease spectrum have been described with mutations in KCNA1, encoding the voltage-gated K+ channel subunit Kv1.1. All of these mutations are either transmitted in an autosomal-dominant mode or found as de novo events.
Methods A patient presenting with a severe combination of dyskinesia and neonatal epileptic encephalopathy was sequenced by whole-exome sequencing (WES). A candidate variant was tested using cellular assays and patch-clamp recordings.
Results WES revealed a homozygous variant (p.Val368Leu) in KCNA1, involving a conserved residue in the pore domain, close to the selectivity signature sequence for K+ ions (TVGYG). Functional analysis showed that mutant protein alone failed to produce functional channels in homozygous state, while coexpression with wild-type produced no effects on K+ currents, similar to wild-type protein alone. Treatment with oxcarbazepine, a sodium channel blocker, proved effective in controlling seizures.
Conclusion This newly identified variant is the first to be reported to act in a recessive mode of inheritance in KCNA1. These findings serve as a cautionary tale for the diagnosis of channelopathies, in which an unreported phenotypic presentation or mode of inheritance for the variant of interest can hinder the identification of causative variants and adequate treatment choice.
- KCNA1
- neonatal epileptic encephalopathy
- dyskinesia
- Kv1.1
- WES
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Episodic ataxia 1 (OMIM: #160120) is a dominantly inherited disease caused by missense variants in KCNA1, encoding the voltage-gated potassium channel subunit Kv1.1. The pathogenic causative variants may appear as de novo events or cosegregate in several affected generations.1 The classical phenotype is defined by myokimia and periodic, intense ataxia episodes accompanied by spastic contractions.2 Patients with mutated KCNA1 present with a wide range of clinical conditions, including epilepsy,3–5 neuromyotonia,6 migraine,7 cerebellar dysfunction with cognitive delay,8 hypomagnesaemia,9 malignant hyperthermia,10 cataplexy,11 contractures12 or paroxysmal kinesigenic dyskinesia,13 among others. Thus, clinical heterogeneity is strong, even between patients carrying the same pathogenic variant.2
Kv1.1 is a member of the voltage-gated, Shaker-related subfamily of potassium channels. These channels regulate neuron excitability by contributing to the resting membrane potential and allowing repolarisation following an action potential. Kv channels are composed of four α subunits. Each subunit contains six transmembrane segments (S1–S6): the first four (S1–S4) comprise the voltage-sensor domain and S5, S6 and the S5–S6 linker form the pore region. Therefore, a Kv pore is formed by S5–S6 domains belonging to four different subunits.14 Each pore domain contains the conserved sequence Thr-Val-Gly-Tyr-Gly (TVGYG), termed the ‘selectivity filter’, as it enables potassium channels to be extremely selective for potassium ions.15 Kv channels in the nervous system are mainly composed of Kv1.1, Kv1.2 and Kv1.4 subunits, which coassemble into homotetrameric or heterotetrameric channels. Described KCNA1 mutations result mostly in variable loss-of-function effects of the Kv1.1 channel, mainly classified into: (i) an impairment in protein expression, assembly or trafficking to the cell membrane or (ii) alterations in channel function, such as reduced channel conductance, shifts in voltage-dependent activation or altered kinetics.2 16 17 Interestingly, given the heteromeric nature of potassium channels, mutated Kv1.1 subunits can alter the function of other subunits.2 As single mutations can exert an effect on various channel properties and because of genetic and environmental modifier factors, no strong genotype-phenotype associations have been established in these disorders.
In this study, we uncover a novel variant in KCNA1 segregating for the first time in an autosomal-recessive inheritance pattern, in a patient showing a severe, previously unreported, complex phenotype characterised by infantile-onset dyskinesia and neonatal onset developmental, epileptic encephalopathy. Functional testing indicated that this variant results in channel loss-of-function in the homozygous state only.
Materials and methods
Informed consent was obtained from all participants in this study according to the Declaration of Helsinki. The research project was approved by the Clinical Research Ethics Committee for Research Ethics Committee of the Bellvitge University Hospital (PR076/14).
Molecular studies
Genomic DNA was extracted from peripheral blood using standard methods. Whole-exome sequencing (WES) was performed on patient DNA samples using the SureSelect XT Human All Exon V5 50 Mb kit (Agilent) for DNA capture and sequencing with the HiSeq 2000 Platform (Illumina) at CNAG (Centre Nacional d’Anàlisi Genòmica, Barcelona). We prioritised non-synonymous coding variants that had a frequency lower than 0.01 in the ExAC, 1000 genomes and EVS databases. Missense variants were evaluated using several predictors (PolyPhen2, SIFT, Mutation Taster, Meta-SVM). Candidate variants were validated and tested for cosegregation in all available family members by Sanger sequencing.
Sequence alignment was performed using ClustalOmega (https://www.ebi.ac.uk/tools/ msa/clustalo/), with sequences extracted from NCBI. 3D models of KCNA1 wild-type or carrying the p.Val368Leu mutation were obtained using the SWISS-MODEL utility in the ExPASy webpage (https://swissmodel.expasy.org/, 5wie.1.B template). Structures were visualised with PyMOL (https//www.pymol.org).
p.Val368Leu mutation was introduced into the p513-KCNA1 vector, which contains the full-length wild-type KCNA1 cDNA,18 using the QuikChange site-directed mutagenesis kit (Agilent Technologies). KCNA1 full-length cDNA (WT and p.Val368Leu) was amplified and cloned into the pEGFP-N3 vector (Clontech) using the InFusion HD Cloning Kit (Takara Bio), to generate two KCNA1-EGFP fusion vectors in which EGFP (Enhanced Green Fluorescent Protein) was fused downstream of KCNA1. All constructs were checked by Sanger sequencing.
For immunofluorescence analysis, HEK293T cells were seeded into 6-well plates to 90% confluence in 24 hours. Transfections were performed using 1 µg of vector (p513-KCNA1 or pKCNA1-EGFP, wild-type or mutated) and 5 µl Lipofectamine 2000 per well (Life Tech.). After transfection, cells were cultured for 48 hours and then fixed in 4% paraformaldehyde. Cells transfected with p513-KCNA1 vectors were incubated overnight at 4°C with primary antibody anti-Kv1.1, clone K20/78 (1:50; NeuroMab) and for 1 hour at room temperature with secondary antibody Goat Anti-Mouse IgG Alexa Fluor 647 A-21236 (1/1000, Dako). DAPI was used to stain cell nuclei. Confocal microscopy images were acquired with a Leica TCS SL laser scanning confocal spectral microscope using a 63× objective.
Electrophysiological studies
HEK293T cells (100 000 cells) were transiently transfected using the Neon Transfection System (Life Technologies). The pCG-GFP plasmid (0.25 µg) was cotransfected with p513-KCNA1 constructs (1.6 µg in p513-Kv1.1WT (wild-type) and p513-Kv1.1p.Val368Leu or 0.8 µg of p513-Kv1.1WT +0.8 µg of p513-Kv1.1p.Val368Leu to mimic a heterozygous condition). After transfection, cells were transferred to coverslip fragments coated with poly-L-lysine. Electrophysiological recordings were performed 24–48 hours after transfection using the whole-cell configuration of the patch-clamp technique as adapted in our laboratory.19 Low-resistance electrodes (1.5–2.5 MΩ), capacity compensation and subtraction of linear leakage and capacity currents were used. Compensation of series resistance was <60% in all experiments. The holding potential was −80 mV. The bath solution was composed of 138 NaCl, 5.4 KCl, 1 CaCl2, 1.2 MgCl2, 10 glucose and 10 HEPES (in mM), pH 7.4; for the solution in the pipette and inside the cell, 100 K-glutamate, 20 K-fluoride, 20 KCl, 1 Na2-ATP and 10 HEPES, pH 7.2. In the 70 K external solution, 70 mM NaCl was replaced with 70 mM KCl. All experiments were conducted at room temperature (20°C–25°C). Voltage-clamp recordings were obtained with an EPC-10 patch-clamp amplifier using standard protocols designed with Pulse software (Heka Elektronik). Data were filtered at 10 kHz, digitalised at a sampling interval of 20 μs and stored on a Macintosh computer. Off-line analysis of data was performed using Pulse Fit (Heka Elektronik). Current densities were obtained by normalising the current amplitude to the cell membrane capacitance. To obtain the conductance versus voltage curves, the amplitude of the tail currents, recorded in the presence of the 70 K external solution, was normalised to the maximum current and plotted as a function of the pulse potential. The curves reflect the best fits to the averaged current-voltage data points, according to the Boltzmann equation: I=Imax/(1 + exp((V – V1⁄2)/k)), where I is the current measured at each test potential (V); Imax is the maximal current; V1⁄2 is the half-maximal activation voltage; k is the slope factor.
Statistical analysis
Data are expressed as the mean±SEM. Statistically significant differences were evaluated with Student’s t-test, with a threshold set at p<0.05.
Results
Index patient II-1 (figure 1A), a 6-year-old girl, third daughter of healthy, consanguineous parents of Moroccan origin, presented with seizures on the 19th day of life, consisting of tonic posture with impaired consciousness, desaturation and cyanosis. She was treated with phenobarbital with partial control. Metabolic screening (blood, urine and cerebrospinal fluid), karyotype, brain MRI, electromyography and a nerve conduction velocity test yielded normal results. Online supplementary video-electroencephalography showed a slow and disorganised background with occipital paroxysmal activity. She developed weekly tonic-clonic seizures with impaired consciousness, usually triggered by febrile episodes or infections, and refractory to phenobarbital and valproic acid. Very good seizure control was observed after introduction of the sodium channel blocker oxcarbazepine. At 3 years old, she presented generalised choreo-dystonic movements associated with facial grimacing that worsened with stress and anger. At the last follow-up in March 2019, she showed impairment of gross motor skills, walking with support or use of a wheelchair, although she could sit normally (Gross Motor Function Classification System (GMFCS) grade III). She handled objects with difficulty (Manual Ability Classification System (MACS) grade III). She also exhibited severe intellectual disability with a lack of communication and expressive language and behavioural problems.
Supplementary video

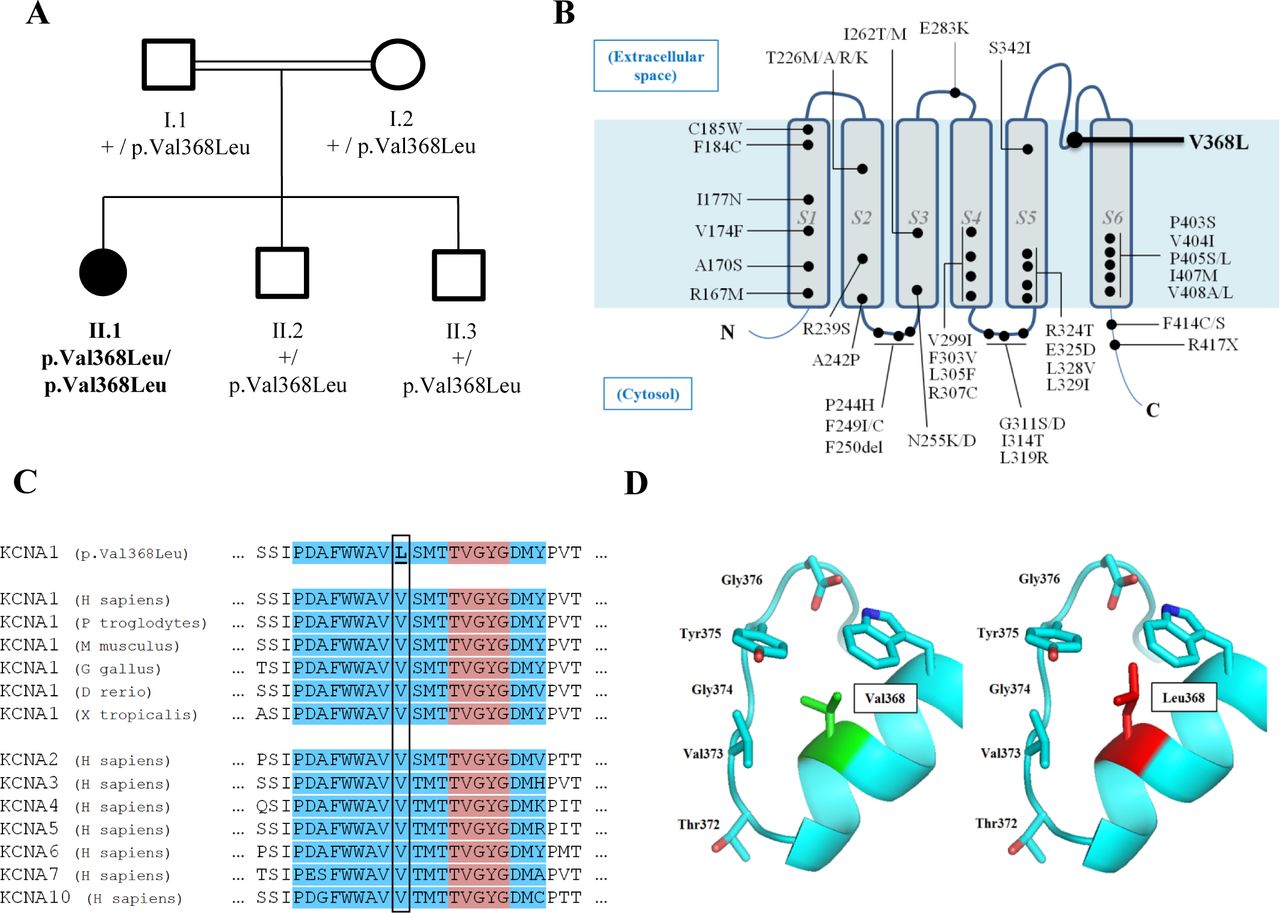
Family tree and KCNA1 p.Val368Leu variant features. (A) Family tree. Square: male, circle: female. Solid black symbols: affected individual. White symbols: unaffected carriers. (B) A representation of Kv1.1 protein in the cell membrane, showing all mutations identified up to 2019 (updated and adapted from2). The position of the p.Val368Leu mutation in the pore region is indicated in bold. (C) Amino acid sequence alignment of Kv1.1 subunits across several species and other members of the human Kv1 family demonstrates conservation of the Val368 residue. Blue indicates the pore intramembrane portion and red indicates the conserved sequence motif functioning as a selectivity filter (TVGYG). (D) Structural models of wild-type and p.Val368Leu KCNA1 sequences. TVGYG, Thr-Val-Gly-Tyr-Gly.
Singleton WES analysis in patient II-1 revealed 12 homozygous extremely rare variants in regions of loss of heterozygosity, compatible with this family consanguineous nature. Among these variants, a missense variant in the KCNA1 gene, p.Val368Leu (Chr12:5 021 646G>C GRCh37, NM_000217:c.1102G>C) was considered a plausible candidate given the phenotypic presentation of the patient. No other candidate rare variants compatible with other modes of inheritance or variants in genes that would remotely relate to the phenotypic features were detected. This variant is completely absent from control databases (1000 Genomes, ExAC, gnomAD). Although KCNA1 has only been associated with dominant conditions (mainly with episodic ataxia/myokymia syndrome, OMIM #160120) in more than 50 families and 260 patients described so far, this variant cosegregated in a recessive mode of inheritance, as all other family members were heterozygous healthy carriers, without a family history of ataxia, dystonia, myokimia or migraine (figure 1A). Although some mutations have been identified in the pore region, this is the first variant reported in the S5-S6 linker (figure 1B). This residue is well conserved throughout evolution (figure 1C), and in silico tools predict the amino acid change to be deleterious. Alignment of human Kv1 channels also indicates that the Val368 residue is conserved among all members of this protein family. Homology models predict this amino acid to directly interact with the conserved signature sequence, TVGYG, which functions as a selectivity filter to conduct K+ ions (figure 1D).14 15 20 Applying ACMG/AMP (American College of Medical Genetics/ Association for Medical Pathology) guidelines for variant interpretation,21 we attributed a variant of unknown significance status to this amino acid change.
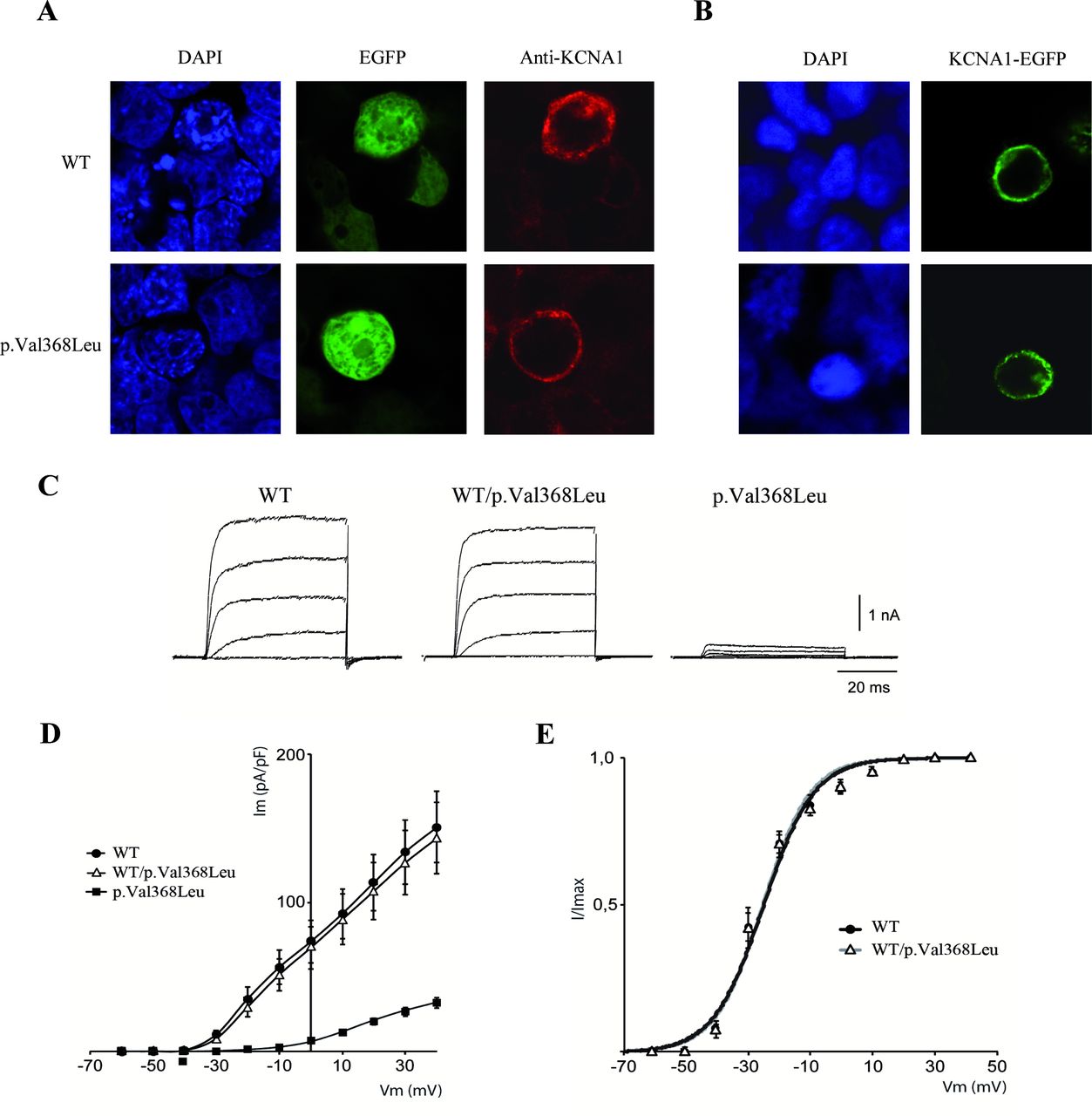
Intrigued by a potential novel inheritance mode, a distinct phenotype for a KCNA1-related disorder, we cloned wild-type and mutant cDNA sequences into expression vectors to assay for localisation. On transfection in HEK293T cells, p.Val368Leu mutant protein localised to the cell membrane in a way similar to the wild-type protein, both using anti-Kv1.1 immunofluorescence or KCNA1-EGFP fusion vectors, implying the lack of trafficking or expression defects conferred by the variant (figure 2A and B). To test the effects of the p.Val368Leu variant on Kv1.1 channel function, we expressed the wild-type protein, the mutant, and a combination of both in HEK293T cells. Cells expressing wild-type Kv1.1 (n=14) showed voltage-dependent potassium currents with a peak current density of 150±24 pA/pF at +40 mV (figure 2C and D). To mimic the heterozygous condition, we injected equivalent amounts of wild-type and Kv1.1p.Val368Leu cDNAs and found currents almost identical to those of wild-type Kv1.1 (peak current density of 143±23 pA/pF at +40 mV, n=15). In contrast, expression of Kv1.1p.Val368Leu yielded significantly smaller currents (peak current density of 35±4 pA/pF at +40 mV, n=15) indistinguishable from the negative control (mock transfected, not shown) or endogenous HEK293T potassium currents. Thus, we conclude that the Kv1.1p.Val368Leu mutant does not produce functional channels. To test whether the coexpression of the p.Val368Leu subunit could affect the voltage dependence of the wild-type Kv1.1, we analysed the conductance vs voltage relationship of the wild-type and heteromeric wild-type/p.Val368Leu channels. No statistically significant differences were observed in the voltage dependence of activation (figure 2E and online supplementary table 1) or in the activation/inactivation kinetics (data not shown) between wild-type and heteromeric Kv1.1 channels.
Supplemental material

{kind=link}
{kind=link}
Functional testing of KCNA1 p.Val368Leu recessive mutation. (A) Immunofluorescence of HEK293T cells transfected with p513-KCNA1WT and p513-KCNA1p.Val368Leu. Blue: DAPI. Green: pEGFP. Red: KCNA1 (anti-Kv1.1 antibody). (B) Immunofluorescence of HEK293T cells transfected with KCNA1WT-EGFP and KCNA1p.Val368Leu-EGFP. Blue: DAPI. Green: KCNA1-EGFP. (C–E) Electrophysiological analysis in cells transfected with p513-KCNA1WT (WT, 1.6 µg), p513-KCNA1p.Val368Leu (p.Val368Leu, 1.6 µg) or both (WT/p.Val368Leu, 0.8 µg+0.8 µg). (C) Representative traces of Kv1.1 outward currents evoked in HEK293T cells transiently expressing WT, WT/p.Val368Leu or p.Val368Leu channels. Currents were recorded at various membrane potentials applied for 50 ms, from −60 to +40 mV in 10 mV increments, from a holding potential of −80 mV. For clarity, only currents registered at –40, –20, 0, +20 and +40 mV are shown. (D) Averaged current density-voltage curves for WT, WT/p.Val368Leu or p.Val368Leu channels. (E) Conductance versus voltage relationships for the WT and WT/p.Val368Leu channels. The lines reflect the best fits to the averaged current-voltage data points, according to the Boltzmann equation as described in the Methods. Values are expressed as the mean±SEM.
Discussion
We report a child affected by a severe neurological disease in which WES uncovers a novel mode of inheritance for a KCNA1-associated disorder. Our patient also shows a previously unreported combination of developmental and epileptic encephalopathy and dyskinesia of neonatal onset, a novel clinical presentation and much more severe phenotype than the classical episodic ataxia and myokimia syndromes most frequently associated with this gene.2 Indeed, infantile-onset epileptic encephalopathy with cognitive impairment has been reported in only four patients harbouring heterozygous de novo KCNA1 variants within the Kv-specific PVP motif, which is essential for channel gating.5 22 A similar phenotype has also been described in KCNA2-mutated patients.23
Functional studies of the p.Val368Leu mutation revealed that the mutant channel is expressed and reaches the cell membrane but causes a strong loss-of-function effect. The p.Val368Leu substitution affects a residue that is well conserved through evolution, located behind the selectivity filter of Kv1.1. It has been suggested that Val368 makes hydrophobic contact with Tyr375 residue’s side chain located in the signature sequence (TVGYG) of the selectivity filter, playing an important role in maintaining its structural integrity.20 Consequently, the substitution of Val by Leu (whose side chain is more voluminous) could seriously affect the interaction with Tyr375, destabilising the selectivity filter and preventing K+ efflux through the pore. Indeed, this amino acid position is crucial for efficient channel gating, as a mutation in the equivalent amino acid of the prokaryotic channel KcsA (Glu71Ala) was shown to abrogate this channel’s natural inactivation process.24 In addition, the p.Val438Ala mutation in the pore region of the Shaker channel (analogous to p.Glu71Ala in KcsA) also resulted into altered C-type inactivation, in contrast to several other mutations generated in this channel.25 Another previously published mutation in the vicinity of the selectivity filter also results in the abolishment of ionic currents in the homologous Shaker potassium channel, in line with our results for p.Val368Leu.26 Thus, our results support a severe alteration of the structural integrity of the channel and block in ion conduction caused by the substitution of the Val368 residue in the Kv family.
Patch-clamp studies of the p.Val368Leu mutation indicate that the electrophysiological properties of the heterozygous state are not significantly different from the wild-type condition. Although we cannot fully exclude a dominant negative effect, these results are consistent with a recessive effect of the mutant allele leading to the absence of clinical manifestations in heterozygote carriers and are analogous to electrophysiological results obtained for mutations in KCNA2 23 and recessive mutations in KCNQ1,27–29 in which cotransfection of wild-type and mutated cDNA also lead to currents hardly different from the wild-type-alone condition. Contrary to KCNA1 mutations behaving in a dominant negative way, the presence of one wild-type allele efficiently counters the loss-of-function effect of the mutated p.Val368Leu allele and prevents heterozygous carriers from developing the disease (figure 2). We posit that transfection of p.Val368Leu-mutant cDNA alone produces channels containing solely mutant KCNA1 subunits, which do not allow K+ currents, while cotransfection of wild-type and mutant cDNAs generates channels containing some wild-type subunits for the most part. In a mixed channel situation, the p.Val368Leu-mutant subunits do not significantly interfere with the wild-type subunits, who carry out their function normally, producing currents in the range of wild-type tetramers. While KCNA1 and KCNA2 mutations can give rise to similar epilepsy phenotypes and both have been associated with an autosomal dominant mode, pLI and pRec scores (which measure probability of intolerance to loss-of-function variants in the heterozygous and recessive state, respectively),30 predict that loss-of-function mutations in KCNA1 (pLI: 0.0959; pRec: 0.897) should behave in a recessive way, while in KCNA2 (pLI: 0.871; pRec: 0.129), this probability is reduced. Concordantly, most KCNA1 mutations are missense variants exerting a dominant negative effect which partially abolishes activity of the wild type protein, in contrast to our variant, which does not affect wild-type function2 18 and behaves as a loss of function allele. It is worth noting that some dominant negative mutations in KCNA1 have been associated with epilepsy,3 although less severely than in our case. This is reinforced by the observation that Kcna1 knockout mice only show spontaneous seizures in the homozygous state.31 Interestingly, our patient exhibited seizure control with a sodium channel blocker (oxcarbazepine). Carbazepine compounds are widely used anticonvulsants that lead to the stabilisation of hyper-excited neural membranes, with well-documented effects in a rat model of myokimia, neuromyotonia and epilepsy harbouring a missense mutation in Kcna1 (Kv1.1S309T).32 The antiepileptic effect of oxcarbazepine was also reported in one patient harbouring a dominant negative variant in KCNA1.12 Interestingly, anticonvulsant effects of these compounds may also be attributed to enhanced conductance of potassium channels.33 Thus, this case adds to the bulk of literature showing improved disease management driven by precision diagnosis and underlines the pertinence of exome sequencing as a first-tier test for early-onset epileptic encephalopathies.34 35
In summary, we have uncovered a novel phenotype and mode of inheritance related to a pathogenic loss-of-function variants in the KCNA1 gene. The location of this mutation close to the selectivity filter demands some caution in interpreting variants, also for other channelopathies, for which the clinical presentation or mode of inheritance may not be concordant with previous descriptions.
Acknowledgments
We are indebted to the family members who participated in this study. We thank CERCA Program/Generalitat de Catalunya for institutional support. We also thank Cristina Guilera, Juanjo Martínez, Arti Mistry, and María Rosa Pezzotti for their excellent technical assistance.
References
Footnotes
AC and AP contributed equally.
Contributors EV, AC and AP designed and conceptualised the study. EV, CF, AS, MR, SF, CC and AC performed analysis and interpreted the data. EV, CF, AC and AP drafted the manuscript. All authors critically revised the manuscript, contributed significantly to this work and declare to meet the ICMJE criteria for authorship.
Funding This study was supported by the Centre for Biomedical Research on Rare Diseases (CIBERER) (ACCI14-759), the URDCat program (PERIS SLT002/16/00174), the Hesperia Foundation and the Secretariat for Universities and Research of the Ministry of Business and Knowledge of the Government of Catalonia (2017SGR1206) to AP and Instituto de Salud Carlos III (PI14/00581) (cofunded by European Regional Development Fund. ERDF, a way to build Europe) and la Marató de TV3 (345/C/2014) to CC and AP. EV was funded by a grant from the Ministerio de Economia, Industria y Competividad (Juan de la Cierva programme FJCI-2016-28811). SF was funded by the Instituto de Salud Carlos III (Miguel Servet programme CPII16/00016, cofunded by European Social Fund. ESF investing in your future) and MR was funded by CIBERER.
Competing interests None declared.
Patient consent for publication Parental/guardian consent obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.