Article Text

Abstract
Cornelia de Lange syndrome (CdLS) is a severe genetic disorder characterised by multisystemic malformations. CdLS is due to pathogenetic variants in NIPBL, SMC1A, SMC3, RAD21 and HDAC8 genes which belong to the cohesin pathway. Cohesin plays a pivotal role in chromatid cohesion, gene expression, and DNA repair. In this review, we will discuss how perturbations in those biological processes contribute to CdLS phenotype and will emphasise the state-of-art of CdLS therapeutic approaches.
- cohesin
- Cornelia de Lange syndrome
- gene dysregulation
- genome instability
- therapeutic approaches
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The cohesin complex, composed of four core subunits, SMC1A, SMC3, RAD21 and STAG, forms a ring-shaped structure that encircles chromatin.1 It is evolutionarily conserved from prokaryotic to eukaryotic organisms. Additional factors, such as NIPBL, PDS5, WAPL, HDAC8 and ESCO, finely regulate the activity of cohesin during the cell cycle (figure 1). Cohesin loading onto chromatin is mediated from NIPBL, in association with its molecular partner MAU2.2 3 It occurs in G1 in yeast or at the end of telophase of the previous cell cycle in mammalian cells. ESCO proteins (1 and 2) allow cohesin establishment in the S phase, and PDS5 (A and B) guarantees its maintenance.4–7 WAPL contributes to cohesin dissolution and HDAC8 is necessary for cohesin recycling during the cell cycle.8 9 Though cohesin was first identified for its role in establishing sister chromatid cohesion, which is important for proper chromosome segregation, cohesin is a key regulator in gene expression10–12 and 3D genome organisation.13 14 In fact, cohesin cooperates with the sequence-specific DNA binding protein CTCF to organise the mammalian genome into structural topologically associated domains (TADs), chromatin loops and contact domains.15–19 It is thought that these features compartmentalise genes and bring together distant enhancers with promoter sequences to orchestrate gene expression.20–22 The disruption of TADs and the removal of cohesin or CTCF binding sites result in abnormal DNA domain topology, thus leading to gene expression dysregulation.14 23–25 Cohesin is also essential for genome integrity as it controls fork replication speed,26 27 promotes DNA repair by homologous recombination,28–32 safeguards telomere stability33 34 and recruits proteins involved in G2/M checkpoints.35 36 DNA damage response and gene transcription are intimately associated via cohesin. In fact, in presence of DNA double-strand breaks, cohesin represses transcription in the flanking chromatin.37 38 Because of this role, it is not surprising that cohesin’s pathogenetic variants are frequently found in cancer, including colorectal and urothelial carcinomas, Ewing sarcomas and acute myeloid leukaemia.39–45
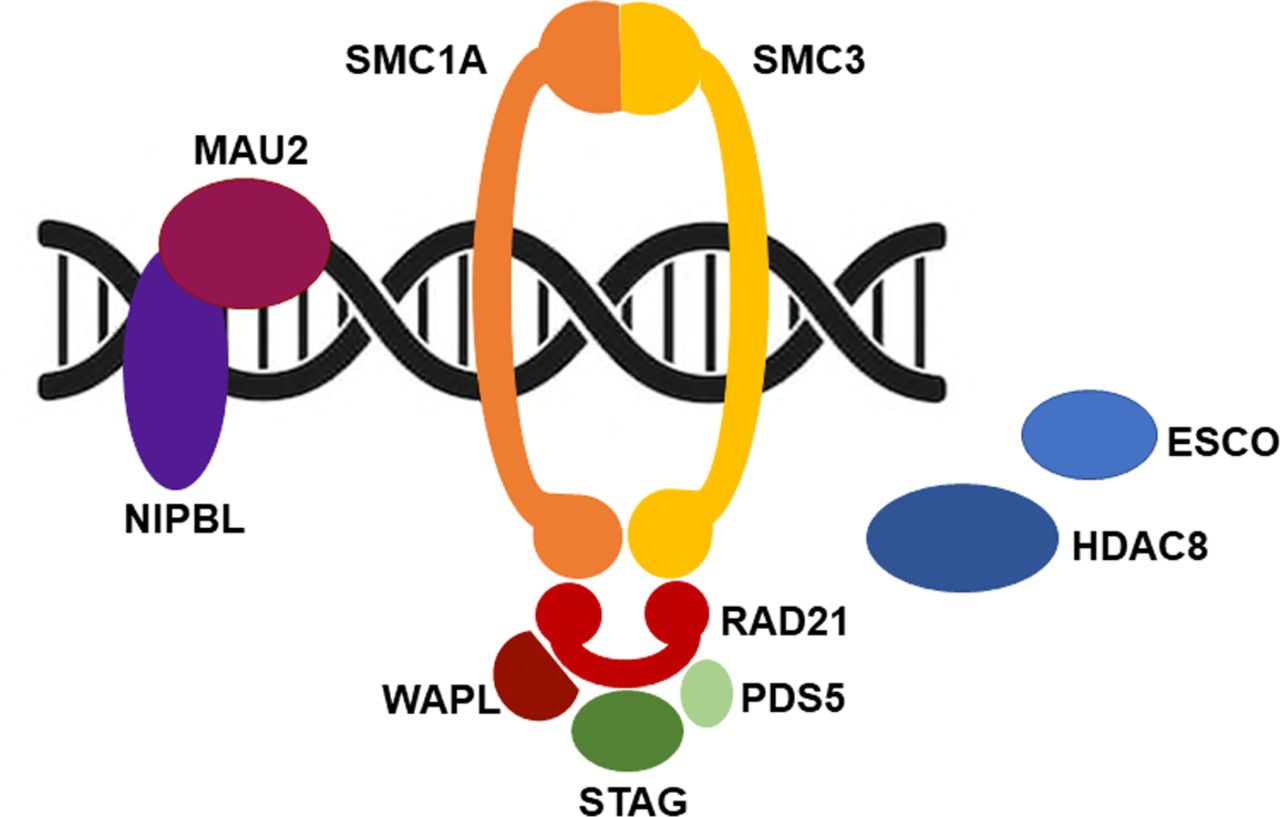
Cohesin and cell cycle. Cohesin is loaded onto chromosomes prior to DNA replication by a binary protein complex formed by NIPBL and MAU2. ESCO (1 and 2) are acetyltransferases that establish sister chromatid cohesion during DNA replication by promoting the acetylation of a pair of lysine residues within SMC3. Pds5 (A and B) has a cohesive effect on cohesin, while having the opposite impact when associated with Wapl. Finally, HDAC8, a histone deacetylase, targets SMC3 permitting cohesin recycling.
Cohesin and Cornelia de Lange syndrome
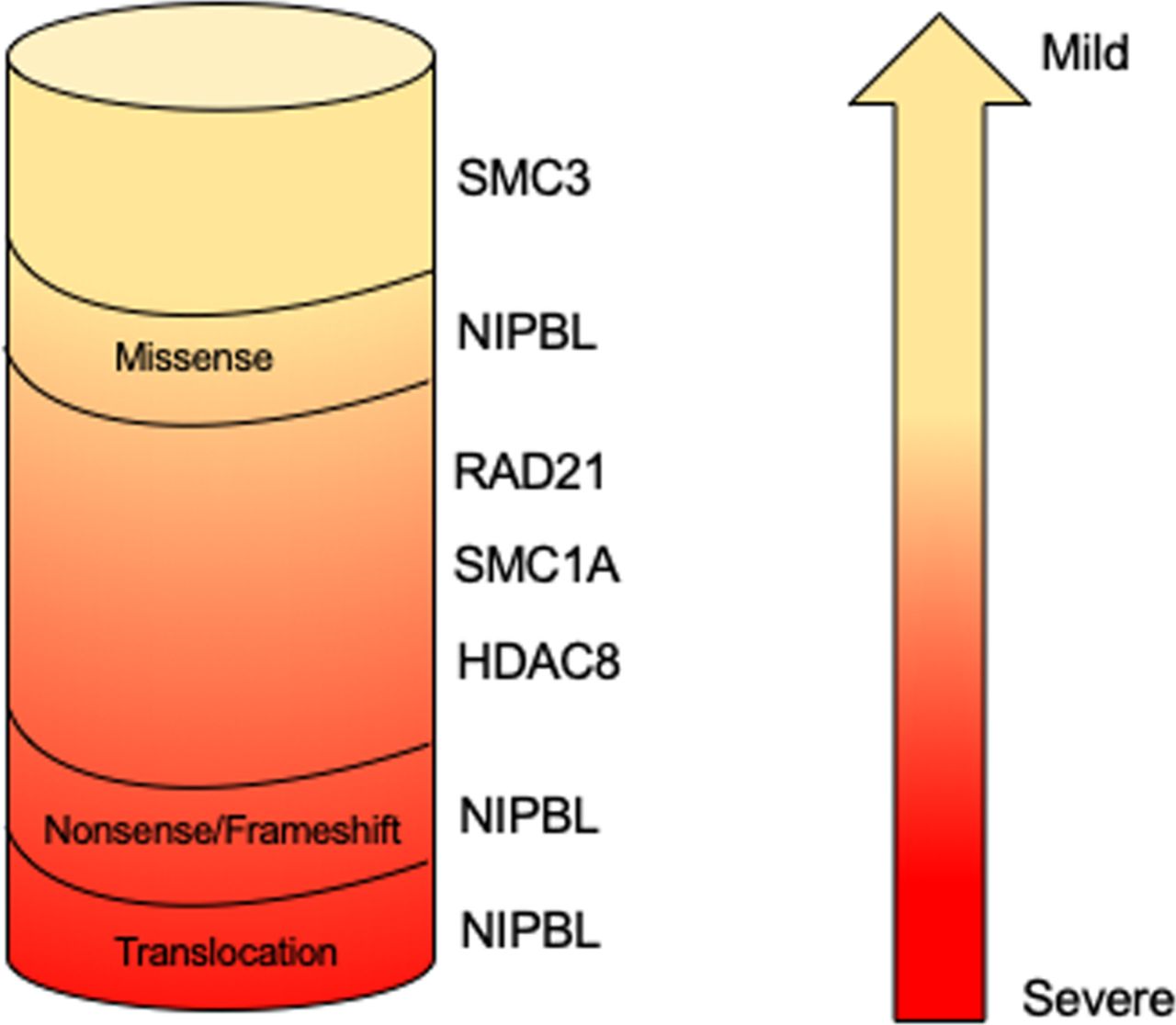
The finding that germinal pathogenetic variants in cohesin structural and regulatory genes are associated with human diseases collectively called ‘cohesinopathies’ may be less counterintuitive. Cornelia de Lange syndrome (CdLS; OMIM 122470, 300590, 610759, 614701, 300882) is the most frequently represented among these. CdLS is a rare multiorgan development disease with a prevalence ranging from 1:10 000 to 1:30 000 live births without differences between ethnic groups.46 Its clinical presentation is characterised by facial dysmorphism (arched eyebrows with synophrys, long philtrum, thin lips, hirsute forehead), prenatal and postnatal growth retardation, cognitive impairment ranging from mild to severe, gastrointestinal malformations, congenital heart abnormalities and limb defects.47 At present, five CdLS-causative genes have been identified. Pathogenetic variants in NIPBL account for about 60% of patients with CdLS whereas about 5%–10% of CdLS probands carry pathogenetic variants in SMC1A, SMC3, RAD21 or HDAC8.9 48–52 This observation suggests that other genes responsible for CdLS have to be identified. Analysis of the mutational spectrum reveals a genotype–phenotype correlation (figure 2). NIPBL truncating, nonsense, splice site and frame shift pathogenetic variants leading to a truncated and likely non-functional NIPBL protein are associated with a severe phenotype. The clinical picture of patients with CdLS carrying SMC1A, SMC3 and RAD21 pathogenetic variants is more uniform, characterised by a mild to moderate phenotype more similar to NIPBL-mutated probands who carry missense changes. Finally, patients harbouring pathogenetic variants in HDAC8 gene show clinical traits overlapping to some extent with classic forms of CdLS characterised by typical facial dysmorphia and severe cognitive delay.53
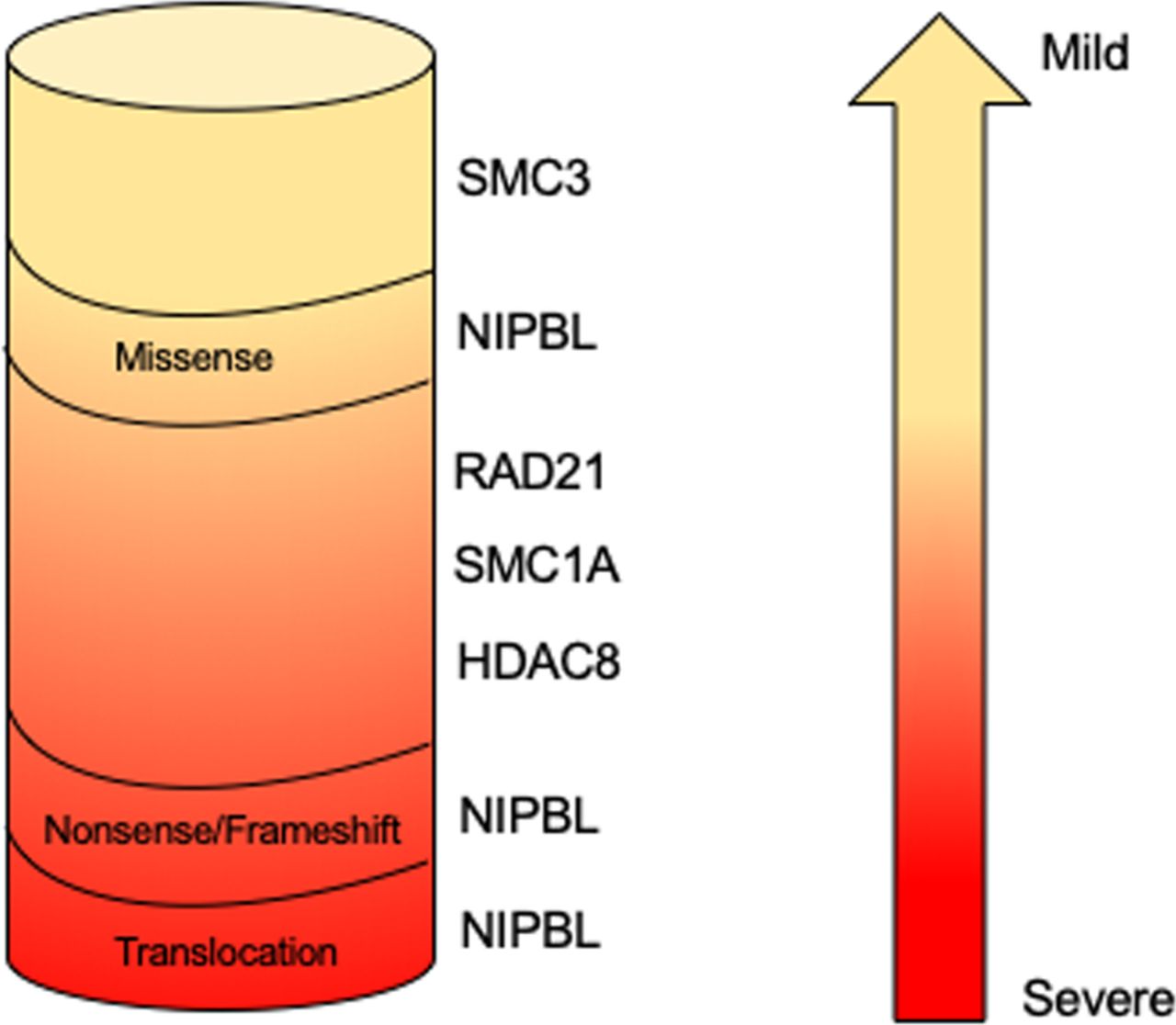
Genotype–phenotype correlation in Cornelia de Lange syndrome. NIPBL truncating pathogenetic variants result in a more severe phenotype, whereas HDAC8, NIPBL, RAD21, SMC1A and SMC3 missense mutations and in-frame deletions are associated with a milder phenotype.
The molecular diagnosis of CdLS is complicated by both the presence of somatic mosaicism and the overlap with other diseases. In fact, sequencing of DNA extracted from fibroblasts or buccal mucosa allowed the identification of mosaic variants in all five CdLS genes in patients originally reported to be mutation negative by Sanger sequencing on DNA isolated from blood.54–58 In order to provide an accurate diagnosis, these variants are of particular interest since they may escape routine molecular diagnostics and indicate that sequencing should not be restricted to DNA from blood samples. Furthermore, patients presenting CdLS or CdLS-like phenotype have been described, and interestingly they carried pathogenetic variants in chromatin-associated factors (table 1) such as BRD4 and AFF4 (responsible for CHOPS syndrome, OMIM 616368); ANKRD11 (associated with KBG syndrome, OMIM 148050); EP300 (responsible for Rubinstein-Taybi syndrome (RSTS), OMIM 613684) and KMT2A (causal gene for Wiedemann-Steiner syndrome (WDSTS), OMIM 605130); and ARID1B, ARID1A, SMARCB1, SMARCA4, SMARCE1, ARID2, SOX11 and DPF2 (associated with Coffin-Siris syndrome (CSS), OMIM 614556, 603024, 601607, 603254, 603111, 609539, 600898, 601671).59–71 Interestingly, all of these disorders display a significant clinical overlap with CdLS, each presenting with shared features that embrace facial dysmorphism, intellectual disability, growth and developmental delay and that are regularly considered in the differential diagnosis of CdLS. These findings suggest that the clinical overlap of these syndromes is mirrored by molecular interactions belonging to the same path, and chromatin dysregulation is a common step towards the pathogenesis of developmental disorders that share phenotypical features with CdLS.
Human diseases overlapping Cornelia de Lange syndrome (CdLS)
Biological processes dysregulated in CdLS
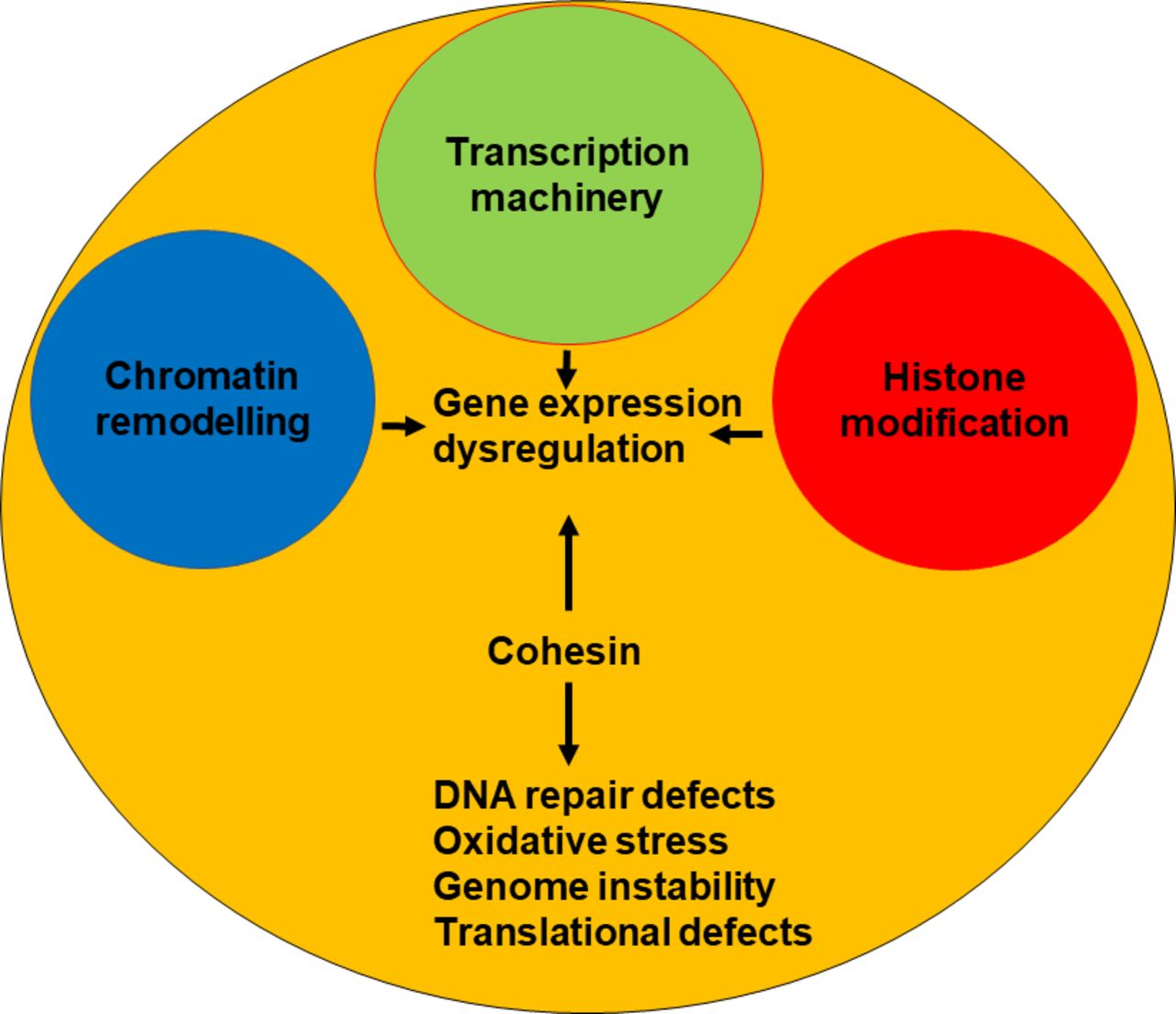
Sister chromatid cohesion is not affected in CdLS72 73; thus, the molecular mechanisms underlying CdLS remain elusive. Several dysregulated biological pathways have been documented in CdLS, including gene transcription, RNA biogenesis, DNA repair and oxidative stress response (figure 3).72 74–78 The cohesin complex, in association with its loader NIPBL and the insulator protein CTCF, regulates enhancer–promoter interaction and is responsible for creating the TADs.18 20 79 Genome-wide data show that cohesin binds more frequently to promoter and downstream regions and is associated with active gene in patients with CdLS.74 75 In addition, CdLS cells harbouring SMC1A pathogenetic variants or the partial decrease in NIPBL expression caused a distinctive profile of gene expression changes.74 75
{kind=link}
{kind=link}
{kind=link}
Most of CdLS are caused by mutations in cohesin and cohesin-regulatory genes. These mutations cause alterations in many fundamental biological processes such as transcription, DNA repair and translation, leading to gene expression dysregulation, high levels of oxidative stress and genome instability. A few cases are instead due to mutations in genes coding transcription machinery members or involved in chromatin remodelling and histone modification, which in turn cause gene changes.
For this reason, CdLS is now grouped in a growing broader class of neurodevelopmental diseases, called disorders of transcriptional regulation (DTRs).80 In addition to CdLS, DTRs include genetic diseases resulting from mutations in gene coding proteins for the transcriptional machinery, histone modification and chromatin remodelling. For example, CSS is characterised by facial dysmorphism, developmental delay, cognitive impairment, absence of terminal phalanges and fifth fingernail hypoplasia.81 CSS is a disorder of chromatin remodelling. In fact, it is caused by mutations in genes belonging to the SWI/SNF chromatin remodeller complex.68 69 CHOPS syndrome is characterised by round faces and arched eyebrows, intellectual disability, heart defects and short stature.59 CHOPS patients carry a mutation in AFF4 gene that encodes a component of the super elongation complex.59 82 Therefore, CHOPS syndrome may be considered a disorder of transcriptional elongation. KBG syndrome is characterised by facial dysmorphism, macrodontia of the upper central incisor, skeletal defects and cognitive impairment.83 KBG syndrome is caused by mutations in ANKRD11 gene which regulates histone acetylation,84 thus it is a disorder of histone modifications. Gene transcription regulation is a fundamental biological process involving many aspects of cell life such as the interaction between enhancer and promoter by chromatin loop, histone modification and the formation of transcription apparatus. The observation that the number of diseases belonging to DTRs is increasing over the years indicates that perturbations in transcription regulation cause multiple developmental syndromes. In addition to gaining insight into the pathogenetic basis of human disorders, the study of DTRs also contributes to dissecting the molecular mechanism of gene transcription regulation.
The findings that CdLS cells show gene expression dysregulation and no defects in sister chromatid cohesion reveal a dosage-sensitive functional hierarchy of cohesin. Mitotic functions appear to be the least dose sensitive, while gene regulation is more dose sensitive. This view is further supported by the findings that the depletion of cohesin or Nipped-B (homologue of human NIPBL) in Drosophila cells and Nipbl in mice affected gene expression but did not cause cohesion or chromosome segregation defects.85 86
The notion that gene transcription is dysregulated in CdLS is further supported by studies in animal models.86–88 Up to 1000 genes have been found dysregulated, but interestingly their fold changes were modest74 75 86 87 suggesting that phenotypic consequences arise from multiple collective perturbations in gene expression. The mechanism leading to gene dysregulation in CdLS is still poorly understood. However, pieces of data may help us to set up the puzzle. In fact, though cohesin is preferentially associated with transcription site start, the number of cohesin sites on differentially expressed genes is significantly reduced in CdLS, whereas the reduction is moderate for the non-differentially expressed genes.74 In addition, mutant cohesin displays an increased affinity for chromatin making the cohesin–DNA binding more stable and impairs PolII recruitment at the promoter regions of dysregulated genes.72 75 It is reasonable to deduce that cohesin mutations cause chromatin modification, leading to global transcription disturbance in CdLS.
Cohesin binds to the ribosomal DNA, plays a role in nucleolus organisation and facilitates protein translation.89 90 It has been shown that rRNA production and protein synthesis are decreased in a zebrafish model of CdLS.91 This finding suggests that cohesin has the potential to affect the functions of the nucleolus, and some of the transcriptional changes observed in CdLS may occur as a result of translational defects.
CdLS cells are characterised by genome instability, as evidenced by the presence of chromosomal rearrangements and aberrations such as translocations, deletions, gaps and breaks.48 49 72 92 In addition, cells are sensitive to genotoxic drugs such as aphidicolin and mytomicin C, and X-ray exposure,72 93 suggesting that CdLS cells have a reduced ability to tolerate DNA damage, likely as a result of reduced DNA repair capability. These data raise the possibility that mutant cohesin may contribute to DNA damage sensitivity by altering the dynamic association of cohesin with DNA, which in turn impairs the recruitment of proteins involved in DNA repair. Oxidative stress could provide a further contribution to the genome instability detected in CdLS. In fact, it is well known that high levels of oxidative stress promote genome instability, apoptosis and cell growth arrest.94–96 Of note, experimental evidence shows the downregulation of proteins involved in the response to oxidative stress and an increase in global oxidative stress in CdLS cell lines.78
Therapeutic approaches in CdLS
The discoveries made in recent years regarding both the identification of CdLS-causative genes and the cellular and molecular characterisation of CdLS cells (figure 3) are the basis for attempting a therapeutic approach in CdLS.
As reported above, CdLS is associated with defects in ribosome biogenesis and translation.91 It has been shown that treatment with l-leucine improves rRNA production, protein synthesis and cell survival and partially rescues developmental defects (embryo length, cartilage formation, and head and eye size) in a zebrafish model of CdLS.91 l-Leucine treatment results in TOR (target of rapamycin) pathway activation through a mechanism that involves the activity of leucyl tRNA synthase and of GTP activating proteins.97 98 The TOR pathway controls cell proliferation, protein translation and ribosome biogenesis.99–101 Treatment with lithium chloride (LiCl) partially rescues neural development in nipblb knockdown zebrafish embryos and cell death in fibroblasts of patients with CdLS.102 Interestingly, LiCl promotes the activation of both Wnt-b-catenin and TOR signalling pathways.103
CdLS is characterised by several phenotypic markers, including growth delay, short stature and delayed puberty.104 Recently, a girl carrying a de novo splicing mutation in NIPBL gene was treated at 4.3 years of age with recombinant human growth hormone (r-hGH). The r-hGH treatment led to a height gain of 1.6 SD score over 8 years105 suggesting that hormonal therapy may be effective for patients with CdLS with short stature.
The relationship between oxidative stress and genome instability in neurodegeneration and senescence is well established.106–108 Oxidative stress is induced by the accumulation of reactive oxygen species. Interestingly, an intriguing link between oxidative stress and TOR pathway has been described, in particular related to age-dependent cognitive decline, pathogenesis of Alzheimer disease and Down syndrome.109–111 Treatment with antioxidant drugs reduces both the level of oxidative stress and genome instability, leading to the extension of in vitro lifespan of CdLS cell lines. In addition, treatment ameliorates the phenotypic feature of zebrafish modelling of CdLS.92 Antioxidant drugs protect the DNA from damage and prevent cell death, as shown in cerebellar cells using N-acetyl-cysteine.112 113 Altogether, these data indicate that antioxidant therapy could provide a means of improving specific phenotypic features of CdLS.
Conclusion
There is increasing evidence that CdLS is caused by a combination of factors, such as gene expression dysregulation, accumulation of cellular damage and cellular phenotype ageing, which collectively contribute to the CdLS phenotype. CdLS therapy is taking its first steps. Chemical or hormonal treatment represents a valuable attempt to identify potential therapeutic targets for future treatment of patients with CdLS, and hopefully clinical trials are now becoming closer.
Acknowledgments
We acknowledge the financial support of Fondazione Pisa to AM.
References
Footnotes
PS and MMP are joint first authors.
Contributors PS and MMP wrote the paper. AM conceived the structure and content, wrote and revised the manuscript.
Funding This study was funded by Fondazione Pisa.
Disclaimer The funder had no involvement.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.