Article Text

Abstract
Background Ambroxol (ABX) has been suggested as an augmentative pharmacological agent for neuronopathic Gaucher disease (nGD). This study assessed the long-term safety and efficacy of combined therapy with high-dose ABX and enzyme replacement therapy (ERT) in nGD.
Methods ABX+ERT therapy was administered for 4.5 years in four patients with nGD. ABX was initiated at a dose of 1.5 mg/kg/day, and the dose was escalated up to 27 mg/kg/day. The target plasma level was 10 µmol/L or less. The changes in glucocerebrosidase activity, biochemical, safety and neurocognitive findings were assessed.
Results Enhanced residual GCcase activity was observed in all patients, as evidenced in both in vitro and in vivo studies. During the first 2 years of study with ABX (up to 21 mg/kg/day), mean seizure frequencies and neurocognitive function worsened. After ABX dosage was increased up to 27 mg/kg/day of ABX, its trough plasma concentration was 3.2–8.8 µmol/L. Drug-to-drug interaction, especially with antiepileptic drug significantly affected the pharmacokinetic parameters of ABX. Importantly, at 27 mg/kg/day of ABX, the seizure frequencies markedly decreased from the baseline, and the neurocognitive function was improved. In addition, Lyso-Gb1, a biomarker for the severity and progression of GD, was normalised in all patients. High-dose ABX was well-tolerated with no severe adverse events.
Conclusions Long-term treatment with high-dose ABX+ERT was safe and might help to arrest the progression of the neurological manifestations in GD.
- metabolic disorders
- Parkinson's disease
- neurology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Gaucher disease (GD; MIM230800) is a lysosomal storage disease caused by glucocerebrosidase (GCase) deficiency. Undegraded substrates, including glucosylceramide (Gb1), its deacylated form (glucosylsphingosine or lyso-Gb1) and other glycolipids, accumulate in the monocyte/macrophage lineage of the reticuloendothelial system, such as the liver, spleen and bone marrow. Accumulation also occurs in the central nervous system (CNS) in some patients causing progressive neurological manifestations, resulting in neuronopathic GD (nGD).1 2
Enzyme replacement therapy (ERT) and substrate reduction therapy (SRT) could achieve significant improvement in the systemic manifestations, including haematological, visceral and skeletal manifestations.3 4 Unfortunately, recombinant enzymes do not cross the blood-brain barrier (BBB) and therefore do not impact on the CNS. The situation with the SRT available today is that both miglustat (despite crossing the BBB) and eliglustat (which is pumped out across the BBB) do not affect the CNS features of nGD, nevertheless—this modality, using different formulations, can lead to changes in nGD, but at present there is only one drug in early phases of clinical trials (venglustat).4–6
Pharmacological chaperones enable the defective enzyme to fold in a manner that restores partial enzyme activity within the lysosome. A well-known mucolytic agent, ambroxol (ABX) has been identified as a pharmacological GCase chaperone,7–10 and six GCase mutant proteins, such as p.Asn227Ser (NP_000148.2) (traditional numbering,11 p.Asn188Ser), p.Phe252Ile (p.Phe213Ile), Gly232Trp (p.Gly193Trp), Arg159Trp (p.Arg120Trp), Gly241Arg (p.Gly202Arg) and Asn409Ser (p.Asn370Ser) have been shown to be responsive to ABX at concentrations of 0.3–30 μmol/L.10 12 Importantly, ABX crosses the BBB and therefore may represent a strategy for mediating therapeutic benefit in the central nervous system.10 13 14 Indeed, previous studies have shown promising results using high-dose ABX in GD. The first study with nGD patients was published by Narita et al 10 in 2016 which tried to verify the effects of ABX in five nGD patients; however, there still remain many uncertainties regarding its long-term safety and efficacy.10 15
In this study, we aimed to investigate the long-term clinical safety and efficacy of combined therapy of high-dose ABX+ERT on the neurological manifestations in patients with nGD.
Methods
Study design
This study was a single-centre study performed at the Asan Medical Center, Seoul, Korea from August 2013 to April 2018. The study protocol was registered at the Clinical Research Information Service, Korea Centers for Disease Control and Prevention, Ministry of Health and Welfare (Republic of Korea) (no. KCT0003218). The detailed information of this study can be found in Clinical Research Information Service (https://cris.nih.go.kr/cris/en/search/search_result_st01.jsp?seq=12387).
The study period was 4.5 years. Four patients (Pt1–4) with nGD were included, and Pt1 and Pt2 were siblings. The baseline clinical characteristics and neurological manifestations of the patients are summarised in table 1. All patients had been treated for 4–14 years with ERT before enrolment (imiglucerase, 60 IU/kg q 2 weeks). ERT dosage was maintained as Abcertin (ISU Abxis, Seongnam, Korea) throughout the study period in all patients. Oral ABX HCl (Mucopect 30 mg; Boehringer Ingelheim, Ingelheim am Rhein, Germany) was administered daily as three divided doses. ABX was started at a dose of 1.5 mg/kg/day, and the dose was escalated every one or 2 months by 3 mg/kg/day up to 21 mg/kg/day or a maximum daily dose of 990 mg until the trough plasma concentration of ABX was 10 µmol/L or less. The trough plasma concentration of ABX was measured three times before increasing its dosage. In addition, cerebrospinal fluid (CSF) ABX concentration was measured when a patient received 21 mg/kg/day of ABX and whenever the dose was escalated. The dosage of 21 mg/kg/day was maintained for 1.5 years and then it was increased up to 1390 mg/day or 27 mg/kg/day and maintained for other 2 years to reach high ABX plasma concentration (10 µmol/L or less).
Baseline characteristics of four patients with neuronopathic Gaucher disease
Efficacy assessment
The primary endpoint in our study was to evaluate the improvement in residual GCase activity in peripheral leukocytes. GCase activity was measured immediately before ERT administration.
Secondary endpoints were the biochemical and clinical outcomes of each patient after the conclusion of the study. Biochemical profiles, such as chitotriosidase, angiotensin-converting enzyme, haemoglobin and platelet count, were measured every 6 months. The frequency of seizures, requirement for antiepileptic drugs, and changes in saccadic eye movement were recorded. The modified severity scoring tool (mSST) and Korean Wechsler adult intelligence scale-IV (K-WAIS) were measured every 6–12 months. The Korean version of modified Barthel index (K-MBI) was measured every 2–2.5 years. Brain magnetic resonance spectroscopy (MRS) was performed every year to evaluate the changes in the metabolite values of N-acetyl-aspartate/creatine (NAA/Cr) and choline/creatine (Cho/Cr) in the frontal white matter and occipital cortex that are known to be affected in nGD.16 17 We also examined the metabolite of the basal ganglia and cerebellum (dentate nucleus), the areas that control exercise and equilibrium functions and where storage cell accumulation has been identified as well.18 Lyso-Gb1 level was also measured in dried blood spots, plasma and CSF, as previously described.19–21 Bone densitometry was performed every year to measure bone density of the lumbar spine and femur neck. Chest X-ray was taken at baseline study and none of the patients had interstitial lung involvement of GD which shows resistance to ERT.
Safety assessment
All adverse events, including clinically significant abnormal findings in the laboratory data, regardless of the severity, were documented and graded using the NCI Common Toxicity Criteria version 4, and it was determined if the adverse event was related to the medical treatment.
Pharmacokinetic (PK) assessment
Blood samples for PK analysis were drawn when the patients received 27 mg/kg/day ABX, before the first ABX (450 mg) dose (0, pre-dose), and at 0.5, 1, 1.5, 2, 3, 4, 5 (the second dose), 6, 7, 8 and 9 hours after the first dose. The serum concentrations were measured by a validated liquid chromatography tandem-mass spectrometry.22 PK of ABX was analysed by a non-compartmental method using WinNonlin software V.6.3 (Pharsight Co, Princeton, New Jersey, USA). The analysis was based on actual sampling time. The peak plasma concentration (Cmax) and the time taken to reach Cmax (Tmax) were determined from observed values. The terminal elimination rate constant (λz) was estimated by linear regression of the terminal log-linear portion of the serum concentration-time curves. The terminal elimination half-life (t1/2β) for each patient was calculated as ln(2)/λz. The area under the serum concentration-time curve (AUC) from time of dosing to 4 hour (AUC0-4h) was calculated by the trapezoidal rule. Based on the AUC and λz, apparent total clearance (CL/F) and terminal volume of distribution (Vz/F), was calculated.
In vivo and in vitro biochemical assays of GCase activity
The GBA (NM_000157) human cDNA clone was obtained (SC120080 OriGene, Rockville, Maryland, USA) and mutant GBA constructs were generated using the PCR-based DpnI-treatment site-directed mutagenesis method. Transient transfection was performed according to the manufacturer’s instructions using Effectene transfection reagent #301 427 (Qiagen, Hilden, Germany). Control primary fibroblasts were obtained from healthy volunteers. GCase enzyme activity assay was performed using a standard fluorometric method.
Fluorescence was detected using a fluorescence spectrophotometer (Molecular devices, San Jose, California, United States). To detect GCcase localisation and cell death and aggresome formation by ABX, control and GD fibroblasts were cultured with or without 10 μmol/L ABX for 5 days. Anti-GCase antibody, MAB7410 (R&D systems, Minneapolis, Minnesota, United States) was added and then LAMP1 antibody, ab24170 (Abcam, Cambridge, UK) was used for lysosome identification. The fluorescent images were acquired using a confocal microscope LSM 780 (ZEISS, Oberkochen, Germany). Aggresome staining was performed according to the manufacturer’s manual (aggresome detection kit; Abcam; ab139486). Positive control cells were treated with 5 µmol/L MG132 for 18 hours. The anti-GCcase antibody (MAB7410) solution was added after staining with the aggresome detection reagent and then the dish was incubated for an appropriate time at dark. The fluorescent images were acquired using a confocal microscope LSM 880 (ZEISS, Oberkochen, Germany). Cell viability was measured by cell counting kit-8 (CCK-8; DOJINDO, CK04, Japan), and cytotoxicity was measured by LDH (lactate dehydrogenase) cytotoxicity detection kit (TAKARA, MK401, Japan). For controls, Triton X-100 (positive control) and untreated normal cells (negative control) were used, and the medium was used for background.
Results
Evidence of enhanced GCase activity by ABX in vitro
The GBA mutations identified in the four patients (Pt1-4) were p.Val211Phefs (traditional numbering,11 Phe171fsX21), p.Asn227Ser (Asn188Ser), p.Phe252Ile (Phe213Ile), p.Arg296Gln (Arg257Gln) and p.Leu483Pro (Leu444Pro).
COS7 cells were transfected with GBA-WT (wild type), p.Asn227Ser, p.Phe252Ile, p.Arg296Gln and p.Leu483Pro DNA and cultured with varying concentrations of ABX (0, 5, 10 and 30 µmol/L) for 2 days.
The residual GCase activity of COS7 cells was significantly enhanced with the increase in ABX concentration, not only in the cells expressing ABX-amendable GBA mutants (p.Asn227Ser and p.Phe252Ile) but also in those expressing p.Arg296Gln and p.Leu483Pro which are unknown GBA variants for ABX responsiveness (figure 1A). Conversely, GCase activity decreased in the cells overexpressing two GBA mutants, p.Arg296Gln and p.Leu483Pro, in the presence of 30 μmol/L ABX.

(A) Glucocerebrosidase (GCcase) activity was measured in COS7 cells transfected with GBA-WT, p.Asn227Ser (traditional numbering,11 Asn188Ser), p.Phe252Ile (Phe213Ile), p.Arg296Gln(Arg257Gln) and p.Leu483Pro (Leu444Pro) at different concentration of ambroxol (0, 5, 10 and 30 μmol/L) for 2 days. (B) GCcase activity was measured in the fibroblasts of four patients with neuropathic Gaucher disease (Pt1–4) at different concentrations of ambroxol (0, 5, 10 and 30 μmol/L) for 5 days. *GCcase activity significantly increased with the increase in ambroxol concentration (p<0.05, compared with the baseline).
Moreover, ABX resulted in a significant enhancement of the residual GCase activity in fibroblasts derived from the four patients (Pt1–4) at different concentrations of ABX (0, 5, 10 and 30 µmol/L) for 5 days (figure 1B). Using confocal microscopy, a focal localisation of GCase in the lysosomes was observed in the fibroblasts of Pt1–4 treated with 10 µmol/L ABX for 5 days (online supplementary figure I), and GCase was not colocalised with aggresome in the juxtanuclear areas (online supplementary figure II). In addition, cytotoxicity was not observed in WT and GD fibroblast cells at 10 μmol/L of ABX, which was assessed by LDH and CCK‐8 assays (online supplementary figure III).
Supplemental material
Evidence of enhanced GCase activity by ABX in vivo
All patients (Pt1–4) completed the study. Dose escalation was uneventful except in one patient (Pt 2), who experienced respiratory difficulty owing to a significant increase in mucus production (online supplementary figure IV). During the first year, the ABX dose was increased by 3 mg/kg/day every 1–2 months up to 21 mg/kg/day or 990 mg/day. The maximum ABX dose was determined based on previous clinical studies using 1000 mg/day.23 24
At a dose of 21 mg/kg/day, the trough plasma concentration of ABX was 2.3–5.7 µmol/L, and the CSF concentration was 0.3–1.4 µmol/L. After maintaining this dose for 1.5 years, the ABX dose was escalated up to 27 mg/kg/day. At this level, the trough plasma concentration of ABX was 3.2–8.8 µmol/L, whereas its CSF concentration was 0.6–1.9 µmol/L. The CSF/plasma concentration ratio was 21.2%±2.9% (figure 2 and online supplementary figure IV).
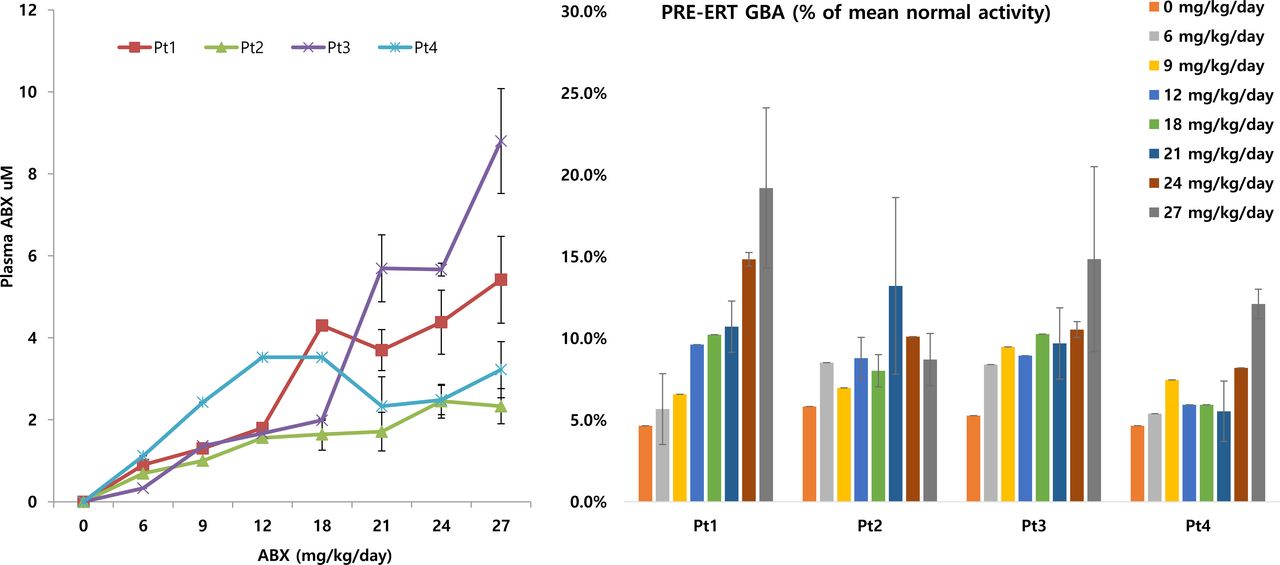
ABX plasma concentration along with the enhancement of residual glucocerebrosidase activity in four patients with neuronopathic Gaucher disease (Pt1–4) receiving combined treatment with high-dose ambroxol and ERT. ABX, ambroxol; ERT, enzyme replacement therapy.
The baseline pre-ERT GCase activity in peripheral leukocytes of Pt1–4 was 5.1±0.6 (range, 4.7–5.8)% of the mean normal activity (ref, 28.7±10.7 nmol/h/mg). At a dose of 21 mg/kg/day ABX, GCase activity was 9.8±3.2 (5.5–13.2)% of the mean normal activity (p=0.042). GCase activity increased up to 13.7±4.4 (8.7–19.2)% of the mean normal activity at ABX dose of 27 mg/kg/day (p=0.020) (figure 2).
Clinical outcomes of nGD after 4.5 years of high-dose ABX+ERT treatment
Biochemical and bone densitometry outcomes
After 4.5 years of high-dose ABX+ERT treatment, no significant change was observed in the haematological profile, such as haemoglobin content and platelet count (online supplementary figure V). All patients had undetectable chitotriosidase activity. The baseline ACE was 54.4±14.1 (33.4–62.6) IU/L, and 31.6±19.3 (5–51) IU/L after 4.5 years. The baseline Z-score of lumbar spine bone densitometry was −0.8±1.3 (-2.5 to 0.5), and −0.5±1.6 (-2.1 to 1.7) after 4.5 years. The baseline Z-score of femoral neck bone densitometry was −0.8±0.4 (-1.2 to −0.2), and −1.0±0.5 (-1.6 to −0.4) after 4.5 years. There were no significant changes in bone densitometry findings during the 4.5 years (online supplementary table I).
Neuropsychiatric outcomes
The mSST score was measured to monitor the neurological progression of GD in each patient (online supplementary table II). At baseline, the mSST score was 12.3±6 (7–20); however, it worsened after 2.5 years of high-dose ABX+ERT treatment (15.1±6.2 (9.5–21)), and then improved after 4.5 years (12.4±5.3 (7–18.5)) (figure 3A). In Pt 1 and 2, improvement of swallowing and speech was noticeable. In addition, Pt 1 showed improvement in pyramidal and extrapyramidal symptoms. Pt 3 and 4 showed improvement of epilepsy and cerebellar tremor.

Changes in modified severity scoring tool (mSST) (A) and Korean version of modified Barthel index (K-MBI) (B) of four patients with neuronopathic Gaucher disease (Pt1–4) during 4.5 years of combined treatment with high-dose ambroxol and enzyme replacement therapy.
The K-MBI score was measured to evaluate daily life movement and performance ability. The mean K-MBI score was 66.3±36.2 (33–100) at baseline. It diminished after 2.5 years (51±32.7 (24–88)) and then improved after 4.5 years (71.8±30.6 (41–100)) (figure 3B). Major improvements were identified in the categories of feeding, dressing, transfer and mobility.
At baseline, the frequencies of myoclonic and generalised tonic-clonic (GTC) seizures were 8.5±14.5 (0–40) times/2 weeks and 5±3.8 (2–10) times/2 weeks, respectively. After 2.5 years, the number of myoclonic seizures decreased to 2±4 (0–8) times/2 weeks; however, the frequency of GTC seizures was 22±30.2 (0–64) times/2 weeks. After 4.5 years, myoclonic and GTC seizures markedly decreased to 1.25±2.5 (0–5) times/2 weeks and 0.5±1 (0–2) times/2 weeks, respectively. Meanwhile, the number of antiepileptic drugs also increased from 4.8±1.7 (3–7) per patient at the baseline to 6.3±2.4 (3–8) per patient after 4.5 years (figure 4 and online supplementary table III).

Changes in seizure frequency and number of antiepileptic drugs (AEDs) in four patients with neuronopathic Gaucher disease (Pt1–4) during 4.5 years of combined treatment with high-dose ambroxol and enzyme replacement therapy.
The K-WAIS score was analysed to evaluate the changes in intelligence. At baseline, the verbal comprehension index (VCI), perceptual reasoning index (PRI), working memory index (WMI), processing speed index (PSI) and full-scale intelligence quotient (FSIQ) were 65±6.8 (56–72), 55±10 (49–70), 60±6.6 (52–66), 50±2.1 (47–52), and 52±6 (46–60), respectively. After 4.5 years of treatment, VCI, PRI, WMI, PSI and FSIQ were 66±7.4 (57–74), 52±3.4 (50–57), 53±3.5 (50–58), 50±0 (50–50) and 48±4.1 (44–53), respectively (online supplementary figure VI).
There was no improvement in horizontal and vertical saccadic eye movement after 4.5 years (online supplementary video). Importantly, it deteriorated in two patients (Pt1 and Pt2) and remained stationary in the other two patients (Pt3 and Pt4). Standing and walking balance was maintained in Pt3 and Pt4 through the study period. In Pt1, it deteriorated until 2.5 years of study and then recovered during the next 2 years. In Pt2, no recovery was observed.
Supplementary video
There were no significant changes in the metabolite values of NAA/Cr and Cho/Cr in the frontal white matter, basal ganglia, occipital cortex and cerebellum (dentate nucleus) as shown by brain MRS (online supplementary figure VII).
Pharmacokinetics
A PK analysis was carried out when all patients were receiving 27 mg/kg/day ABX or 450 mg ABX three times daily.
The Cmax was 0.46±0.19 (0.26–0.72) ng/mL, Tmax was 1.13±0.48 (0.5–1.5) hour, AUC0-4h was 1.68±0.73 (0.96–2.68) hour*ng/mL, CL/F was 34.27×103 ± 9.45×103 (9.45×103–200.14×103) L/h, Vz/F was 1.08×106 ± 0.49×106 (0.6×106–10.75×106) L/h, and t1/2β was 21.7±6.47 (15.56–30.8) hour (online supplementary figure VIII). Since a wide variation in the PK parameters was observed among patients, drug–drug interactions were assessed. ABX is metabolised by CYP3A4,25 and topiramate administered to Pt1, 2, 4 and carbamazepine administered to Pt2 are CYP3A4 inducers, whereas divalproate sodium administered to Pt2, 3 is a weak CYP3A4 inhibitor. In particular, the Cmax and AUC0-4h in Pt3 were higher than those in Pt1, 2 and 4 (online supplementary figure VIII).
Safety assessment
High-dose ABX was generally safe and tolerable in all patients. Several drug-related side effects were observed (online supplementary table IV); however, they did not require drug discontinuation. Pt3 occasionally showed transient proteinuria ranging from 34 to 67.5 mg/day. Mucus production significantly increased in Pt2, who complained of respiratory difficulty; thus, escalation of ABX dose was delayed in this patient. However, ABX dose was slowly escalated up to 27 mg/kg/day in Pt2, and this dose was tolerable.
Changes in Lyso-Gb1
Lyso-Gb1 level was measured as a corollary for the severity and progression of GD.20 As the dose of ABX increased, plasma lyso-Gb1 level decreased in all patients (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Plasma Lyso-Gb1 levels in four patients with neuronopathic Gaucher disease (nGD) (Pt1–4) during 4.5 years of combined treatment with high-dose ambroxol and enzyme replacement therapy. Plasma glucosylsphingosine (lyso-Gb1) level of Pt1-4 receiving 27 mg/kg/day ambroxol was 5.3±2.5 (2.4–11.6) ng/mL. (B) The lyso-Gb1 level of Pt1–4 in the dried blood spots (7.7±1.8 (6.5–10.3) ng/mL) was significantly lower than those of 9 non-nGD patients (93.8±68.4 (27–115) ng/mL) ng/mL and of 3 nGD patients without ambroxol treatment (45.7±19.8 (25.7–77.2) ng/mL) (ref.,<4.8 ng/mL) (p<0.01).
Lyso-Gb1 was also measured in dried blood spots from nine non-nGD patients receiving ERT alone (30–60 IU/kg every other week) for 10.9±8.3 years and from 3 nGD patients receiving ERT alone (60–120 IU/kg every other week) for 4–24 years. Lyso-Gb1 level was 93.8±68.4 (27–115) and 45.7±19.8 (25.7–77.2) ng/mL in the non-nGD and nGD patients, respectively. At ABX dose of 27 mg/kg/day, lyso-Gb1 level in the dried blood spots of Pt1-4 was 7.7±1.8 (6.5–10.3) ng/mL (ref,<4.8 ng/mL), which was significantly lower than that in non-nGD and nGD patients without ABX treatment (Mann-Whitney test, p<0.01) (figure 5). At ABX dose of 27 mg/kg/day, CSF Lyso-GB1 level was 0.52±0.04 ng/mL (9.2%±2.5% of plasma Lyso-Gb1).
Discussion
Long-term treatment with high-dose ABX+ERT was tolerable in all patients during the study duration (4.5 years). Asymptomatic mild proteinuria and increased respiratory mucus production were the only major related adverse reactions. Since the degree of proteinuria is related to renal function, a careful follow-up is required for monitoring proteinuria and renal function. Importantly, the increase in respiratory mucus production was the major adverse event that hindered ABX dose escalation.
Importantly, our in vitro and in vivo data showed significant increases in GCase activity. In addition, an increase in the subcellular localisation of lysosomes was observed in patient-derived fibroblasts treated with 10 μmol/L ABX, indicating that ABX facilitated its trafficking to the lysosomes.
In a previous study by Narita et al, high-dose ABX+ERT showed remarkable improvement in myoclonus, myoclonic seizures and pupillary light reflex dysfunction in five nGD patients despite the variation in the treatment period (0.5–4 years). ABX was administrated and escalated at the target dose of 25 mg/kg/day or a maximum dose of 1300 mg/day. The frequency of myoclonic seizures decreased after increasing the dose of ABX to 9 or 15 mg/kg/day. Frequency of GTC seizures did not change, but the duration of seizure or frequency of status epilepticus decreased in some patients. Moreover, two patients were able to walk again with an impressive recovery of gross motor function.10 However, during the first 2.5 years of our study, the neurological manifestations were not improved or in some cases aggravated, as evidenced from the mSST and K-MBI scores. Considering that all the patients in our study and that by Narita et al both harboured the ABX-amendable mutations, p.Asn227Ser or p.Phe252Ile, except in one patient in the study by Narita et al, the different pattern of response is not considered as related to GBA mutation spectrum. In our study, because the dosage of ABX increased by 3 mg/kg/day every 1–2 months, the natural progression of neurological manifestations might have not been delayed by the therapeutic effects of ABX during the early period of our study.
An improvement in the neurological manifestation was observed only after the dose of ABX increased up to 27 mg/kg/day during last 2 years of the study. The mSST and K-MBI scores were stabilised/maintained, and the neurocognitive function was sustained in all patients. In addition, seizure frequency markedly decreased. Although the number of antiepileptic drugs increased during our study, the remarkable prevention of seizures was meaningful clinically and in quality of life improvement.
The threshold enzyme activity to prevent substrate accumulation has been suggested to be ˃ 10%–15% of normal.26 In our study, approximately 9%–19% of the mean normal GCase activity was achieved in patients who received 27 mg/kg/day ABX (figure 2). Luan et al 12 showed that GCase activity significantly increased in the brains of normal mice receiving 60 mg/kg/day ABX, which is equivalent to a human dose of 4.8 mg/kg/day.27 However, the degree of improvement in brain GCase activity to avoid Gb1 and lyso-Gb1 accumulation is not known. In addition, it is noteworthy that since ABX acts as a chemical chaperone to GCase, it may also act as an inhibitor at high concentrations.8 In support of this, as shown in figure 1A, ABX acted as an inhibitor of some GCase mutants at concentrations of 30 μmol/L and higher. Previous in vitro and in vivo studies indicated that ABX enhanced GCase activity at concentrations of 0.3–30 μmol/L.8 10 12 However, based on our results, there may be impetus to increase the ABX dosage to achieve a plasma concentration of 10 μmol/L. At this concentration, cytotoxicity was not observed as demonstrated by LDH and CCK‐8 assays and aggresome staining. However, the safety and efficacy of higher doses to achieve a plasma concentration of 30 μmol/L needs to be solved on further studies.
In our study, GCase activity was enhanced up to 9%–19% of the mean normal activity at ABX dose of 27 mg/kg/day, which was significantly lower than that reported by Narita et al , where GCase activity was enhanced up to 40%–100% of the normal activity at ABX dose of 25 mg/kg/day.10 One explanation for this discrepancy might be the variability in the PK characteristics among the subjects in our study (figure 5). ABX is metabolised by CYP3A4, and drug–drug interactions between ABX and CYP3A4 inducers or inhibitors can affect ABX concentration and subsequently affect GCase activity. Several antiepileptic drugs, such as topiramate and carbamazepine (CYP3A4 inducers) and divalproate sodium (CYP3A4 inhibitor) can significantly affect ABX concentration. In particular, in Pt2 who received carbamazepine, ABX concentration was persistently low (2.33±0.43 μmol/L) even at a dose of 27 mg/kg/day.
Impairment of saccadic eye movement is one of the characteristic findings in nGD.1 2 In a previous study,10 high-dose ABX was effective in improving pupillary light reflex and latency in saccadic eye movement. In our study, these parameters were not assessed in detail; however, saccadic eye movement deteriorated in two patients (online supplementary video).
Lyso-Gb1, a biomarker for the severity and progression of GD,19 28 gradually decreased in all patients until reaching a level of 7.7±1.8 (6.5–10.3) ng/mL, which is comparable to the normal range (<4.8 ng/mL).29 Moreover, Lyso-Gb1 level in patients treated with ABX+ERT was much lower than that in non-nGD and nGD patients who were treated with long-term ERT without ABX. These data suggest evidence for the protective effects of ABX in GD in vivo.
Collectively, our results show that long-term treatment with high-dose ABX was generally safe. Based on the results of the mSST and K-MBI scores, seizure frequency and Lyso-Gb1 levels, long-term treatment with high-dose ABX abated the progression of neurological manifestations in GD. However, because of the wide variability in plasma ABX levels among patients, assessment of drug–drug interactions (particularly with antiepileptic drugs) will be important to monitor in order to define the optimal dosage of ABX in each patient. Moreover, the clinical severity of the neurological manifestations at baseline in each patient was an important factor affecting the clinical outcome of long-term treatment with high-dose ABX+ERT. Therefore, earlier administration of ABX, even before the progression of the neurological features (such as immediately on beginning of ERT in newly diagnosed nGD patients), might have a greater value and achieve better results. Also, rapid escalation of ABX dose might lead to faster and better outcomes. However, this may be limited by the significant respiratory mucus production and drug–drug interactions.
Recently, GBA mutations have been suggested to be associated with the development of Parkinson’s disease.30 Therefore, the promising results of high-dose ABX treatment in nGD patients suggest that high-dose ABX might be a potential pharmacological therapy for more common neurodegenerative conditions. Our study showed improvement in four patients and supports larger scale studies to better define the dosing parameters for wider spread application in nGD.
Acknowledgments
We deeply appreciate the patients and their families for participating in this study. We acknowledge Professor Kousaku Ohno, Tottori University Faculty of Medicine, Yonago, Japan for his grateful consultation.
References
Footnotes
Y-MK and M-SY contributed equally.
BHL and H-WY contributed equally.
Contributors BHL, HKJ, J-SB, AR, AZ and H-WY contributed to designing the study. Y-MK, M-SY, GHS, AO, HMY, HTL, H-WK, T-SK, BHL and HMY were the clinicians who conducted all clinical and radiological examinations. S-HH, T-SK, H-SL, CC and AR did the laboratory experiments. BHL, Y-MK, M-SY, HKJ, J-SB, H-WY, HTL, MJO, JT, AR, AZ and HMY analysed the data. Y-MK, M-SY, S-HH and BHL drafted the manuscript and HKJ, MJO, JT, CC, AR, AZ revised the manuscript. All authors were involved in analysing and interpreting the data. All authors read and approved the final manuscript.
Funding This research was supported in part by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (NRF-2015K1A4A3046807, NRF-2016M3A9B4915706 and NRF-2018M3A9H1078335) and by ISU ABXIS, Gyeonggi-do, Korea.
Competing interests None declared.
Patient consent for publication Parental/guardian consent obtained.
Ethics approval The study was approved by Institutional Review Board, Asan Medical Center, Seoul, Korea and the Ministry of Food and Drug Safety, Korea.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.