Article Text

Abstract
Introduction Lynch syndrome (LS) and constitutional mismatch repair deficiency (CMMRD) are hereditary cancer syndromes associated with mismatch repair (MMR) deficiency. Tumours show microsatellite instability (MSI), also reported at low levels in non-neoplastic tissues. Our aim was to evaluate the performance of high-sensitivity MSI (hs-MSI) assessment for the identification of LS and CMMRD in non-neoplastic tissues.
Materials and methods Blood DNA samples from 131 individuals were grouped into three cohorts: baseline (22 controls), training (11 CMMRD, 48 LS and 15 controls) and validation (18 CMMRD and 18 controls). Custom next generation sequencing panel and bioinformatics pipeline were used to detect insertions and deletions in microsatellite markers. An hs-MSI score was calculated representing the percentage of unstable markers.
Results The hs-MSI score was significantly higher in CMMRD blood samples when compared with controls in the training cohort (p<0.001). This finding was confirmed in the validation set, reaching 100% specificity and sensitivity. Higher hs-MSI scores were detected in biallelic MSH2 carriers (n=5) compared with MSH6 carriers (n=15). The hs-MSI analysis did not detect a difference between LS and control blood samples (p=0.564).
Conclusions The hs-MSI approach is a valuable tool for CMMRD diagnosis, especially in suspected patients harbouring MMR variants of unknown significance or non-detected biallelic germline mutations.
- lynch syndrome
- constitutional mismatch repair deficiency
- microsatellite instability
- next generation sequencing
- highly sensitive methodologies
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- lynch syndrome
- constitutional mismatch repair deficiency
- microsatellite instability
- next generation sequencing
- highly sensitive methodologies
Introduction
Lynch syndrome (LS; OMIM #120435), the most prevalent hereditary colorectal and endometrial cancer syndrome, is an autosomal dominant cancer-susceptibility disease caused by inactivating heterozygous germline mutations in mismatch repair (MMR) genes (MLH1, MSH2, MSH6 and PMS2).1 Constitutional mismatch repair deficiency (CMMRD; OMIM #276300) is a rare devastating cancer syndrome caused by biallelic germline mutations in the same genes and mainly characterised by the development of haematological, brain and colorectal tumours during childhood and adolescence.2 3 Overlapping phenotypes have been described between LS and CMMRD,4 5 as well as between CMMRD and other cancer syndromes such as neurofibromatosis type 1 (NF1), polymerase proofreading-associated polyposis (PPAP) and Li-Fraumeni.6 7
The identification of these inherited conditions has important consequences for the clinical management of carriers.8 9 Molecular diagnosis of LS and CMMRD is often hampered by the identification of variants of unknown significance (VUS) in about 30% of all identified MMR variants and by difficulties in sequencing PMS2 due to multiple pseudogenes, which accounts for approximately 60% of CMMRD cases.3 6
In LS, somatic inactivation of the MMR wildtype allele initiates an accumulation of errors mainly in repetitive sequences. Consequently, LS-associated tumours are hypermutated (>10 mutations/Mb), exhibit microsatellite instability (MSI) and lose expression of MMR proteins.1 In CMMRD, the germline inactivation of both MMR alleles together with somatic polymerase exonuclease domain mutations leads to ultra-hypermutated tumours (>100 mutations/Mb).10 The CMMRD diagnostic hallmark is the loss of MMR protein expression in both tumour and normal tissue.3 7 However, some missense mutations are associated with conserved expression and MSI may be negative in CMMRD tumours, especially in non-gastrointestinal cancers.2 3
Besides the recently reported in vitro repair assay in lymphocytes,6 tools have been developed to assess the degree of MSI in CMMRD normal tissues. The germline MSI (gMSI) assay,11 based on electropherogram analysis of three dinucleotide markers, has demonstrated high specificity but low sensitivity due to its inability to identify biallelic MSH6 mutation carriers. The ex vivo MSI analysis,12 based on lymphoblastoid cell lines, in combination with a methylation-tolerance assay, showed higher sensitivity for CMMRD identification. Recently, a next generation sequencing (NGS) approach to detect gMSI has shown high accuracy.13 None of these techniques are sensitive enough to detect MSI in normal tissues from LS carriers. Nevertheless, low-level MSI has been reported in blood DNA from individuals with LS using laborious single-molecule analyses.14 15 Notably, MMR deficiency has been detected in apparently normal colonic and endometrial epithelium of LS carriers.16
We hypothesised that an assessment of MSI markers at high sensitivity could improve the diagnosis of cancer syndromes associated with MMR deficiency. Our aim was to evaluate the performance of high-sensitivity MSI (hs-MSI) assessment in normal tissues of LS and CMMRD carriers.
Materials and methods
Patients and samples
Samples from 131 individuals were grouped into three cohorts: baseline, training and validation. The baseline cohort comprised 22 healthy control samples; the training cohort included 74 blood samples from healthy controls, patients with CMMRD and individuals with LS (online supplementary table S1); and the validation cohort comprised 36 blinded samples from individuals with clinical diagnosis of CMMRD3 and healthy controls, kindly provided by the European Consortium C4CMMRD (online supplementary table S2). Some samples were also analysed in a recent study13 (online supplementary tables S1 and S2). An oral mucosa sample from a patient with CMMRD (online supplementary table S1), four cases with CMMRD-suspected diagnosis and mutation carriers of CMMRD overlapping syndromes were also included (online supplementary table S3). Five DNA samples from frozen tumours were used as controls, two classified as MSI and three as microsatellite stable (MSS), using the MSI Analysis System (Promega). Genomic DNA was obtained using standard protocols.
Supplemental material
Assessment of MSI at high sensitivity (hs-MSI)
The analytical sensitivity of variant detection by using a molecular barcoding-based NGS approach was initially assessed with the ClearSeq Cancer HS panel (Agilent Technologies; online supplementary methods).
Supplemental material
A custom panel targeting 277 microsatellites, 91% of them mononucleotide repeats, was designed using HaloPlex HS technology (online supplementary figure S1, online supplementary methods). Sequencing of enriched regions was performed in a HiSeq platform at high coverage (20 000×), reaching a mean depth of 1312±447 reads/marker/sample after deduplication. A set of 231 truly monomorphic microsatellites in the baseline were selected. Among them, 186 markers were previously reported as frequently mutated in tumours with high instability (MSI-H). A bioinformatics pipeline for microsatellite indel calling was customised (online supplementary figure S2, online supplementary methods).
Supplemental material
To assess the hs-MSI status at each microsatellite locus, the instability level, corresponding to the sum of the frequencies of all allele lengths different from the wildtype (mutational load method), was calculated as (1 – wildtype allele frequency). Alternatively, the frequencies of each alternative microsatellite allele length were used (individual allele method). Whenever the instability level or frequency of alternative allele exceeded the mean value in baseline plus 3 SD and the highest value among the individual samples of the baseline, the microsatellite was considered unstable.
For both methods, an MSI score was calculated per sample, representing the percentage of unstable markers. hs-MSI median score was compared between different training set groups using a Wilcoxon rank-sum test (online supplementary figure S2, online supplementary methods).
Analysis of dinucleotide repeats
gMSI analysis of the dinucleotide markers D17S791, D2S123 and D17S250 was performed as described.11 Analysis of D2S123 from NGS data was described in online supplementary methods.
Results
The percentage of unstable monomorphic markers frequently mutated in MSI-H tumours included in the hs-MSI panel (n=186; mutational load method) was higher in the DNA from MSI-H than MSS colorectal tumours (online supplementary figure S3A). This MSI score was significantly higher in blood DNA samples from patients with CMMRD (median=23.58%) compared with healthy controls (median=1.10%) (p=1.24e-05) or LS blood samples (median=0.85%) (p=9.49e-08), without overlapping (figure 1A and online supplementary table S1). No evidence of clonal expansion was seen in haematological CMMRD samples. In contrast, no difference was detected between LS and control samples (p=0.564) (figure 1A and online supplementary table S1). Similar results were obtained using the whole set of monomorphic markers (n=231) and when an individual allele method was used irrespective of the absolute values of the thresholds for MSI detection in blood (online supplementary figure S3).

hs-MSI analysis in the training and validation cohorts. Monomorphic microsatellite markers frequently mutated in MSI-H tumours (n=186) analysed using the mutational load analysis method. (A) MSI score in blood DNA samples from LS (median=0.85, IQR=0.55–1.65, range=0.00–3.33), CMMRD (median=23.58, IQR=21.33–25.49, range=14.84–59.22) and healthy individuals (median=1.1, IQR=0.54–1.65, range=0.00–3.89) from the training set. Significant differences were observed between patients with CMMRD and negative controls (***p=1.24e-05), while no differences were found between patients with LS and negative controls (ns, non-significant, p=0.564). Dashed line indicates the threshold for hs-MSI detection in blood samples. (B) MSI score in blinded samples from the validation cohort. Patients with CMMRD (median=26.28, IQR=19.14–38.37, range=10.56–76.50) and negative controls (median=0.57, IQR=0–1.11, range=0–1.79) were discriminated with no overlapping (hatched area) (***p=2.784e-07). Dashed line indicates the threshold for hs-MSI detection. CMMRD, constitutional mismatch repair deficiency; hs-MSI, high-sensitivity microsatellite instability; LS, Lynch syndrome.
Using an independent blinded set of blood samples, the MSI score accurately distinguished patients with CMMRD (median=26.28%) from controls (median=0.57%) (p=2.784e-07) (figure 1B, online supplementary table S2). In this context, the hs-MSI approach displayed a specificity of 100% (95% CI 89.42% to 100%) and a sensitivity of 100% (95% CI 88% to 100%) (online supplementary table S4). In agreement with the results obtained in the gMSI assay, instability at D2S123 dinucleotide marker was detected in biallelic MMR carriers except for MSH6 (online supplementary tables S1, S2 and S5).
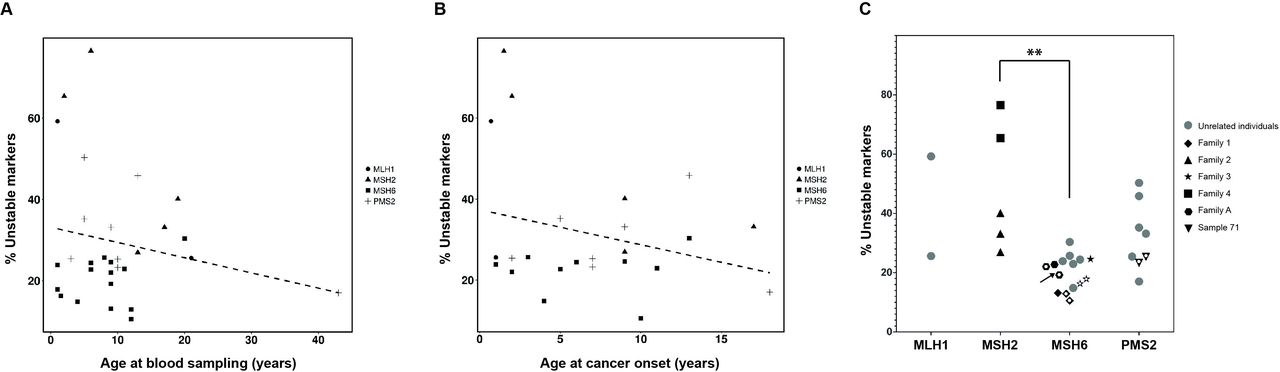
No correlation between MSI score and age at blood sampling was observed in control, LS or CMMRD samples (figure 2A, online supplementary figures S4 and S5). Moreover, no correlation with age of cancer onset was noted in CMMRD (figure 2B) or LS-affected patients (online supplementary figure S5). In contrast, when CMMRD samples were grouped by germline-affected gene, significant differences were observed between instability levels of MSH6 and MSH2 biallelic carriers (p=0.0014) (figure 2C). Furthermore, no dependency of MSI levels and germline affected gene was observed in LS samples (p=0.0523) (online supplementary figure S5A).
{kind=link}
{kind=link}
Characterisation of the hs-MSI observed in CMMRD samples. Monomorphic microsatellite markers (selected as frequently mutated in MSI-H tumours) have been analysed (n=186). (A) MSI score in CMMRD blood samples plotted against patient age at blood sampling. No correlation was observed (dashed line, r=−0.04, p=0.823). (B) MSI score in CMMRD blood samples plotted against age of cancer onset. No correlation was observed (dashed line, r=−0.15, p=0.491). (C) MSI score in CMMRD samples plotted against the germline mutated MMR gene. Samples from the same family are indicated by the same symbol. White dots inside symbols indicate samples from the same individual. The buccal mucosa sample is indicated by an arrow. Statistically significant differences between affected genes are indicated (**p<0.005). CMMRD, constitutional mismatch repair deficiency; hs-MSI, high-sensitivity microsatellite instability; MMR, mismatch repair; MSI-H, tumours with high instability.
An oral mucosa DNA sample (CMMRD-01) displayed similar MSI score to a paired blood sample (figure 2C and online supplementary table S1). Conversely, high hs-MSI score was not detected in the blood from germline TP53, POLE/POLD1 and NF1 mutation carriers, early-onset LS or four cases with a suspected but unconfirmed diagnosis of CMMRD, pointing to the absence of CMMRD in the latter (online supplementary table S3, online supplementary figure S6).
Discussion
Accurate and prompt diagnosis of CMMRD is essential for therapeutic decisions and surveillance recommendations.9 Here we report the performance of the novel hs-MSI approach for high-sensitivity gMSI assessment. Our hs-MSI approach based on the analysis of mononucleotide repeats demonstrated higher accuracy to discriminate between controls and CMMRD cases (including MSH6 biallelic carriers) than previously reported methods,11–13 requires low DNA input (less than 100 ng), and have an estimated turnaround time of 1 week (online supplementary table S6). In addition, the result obtained with a CMMRD individual’s oral mucosa sample suggests its potential for the analysis of MSI in minimally invasive samples, patients with lymphopenia or after allogenic bone marrow transplant. Moreover, the hs-MSI approach is able to robustly discriminate between CMMRD and LS, Li-Fraumeni, NF1 and PPAP, which may assist in classifying cases with overlapping phenotype.4 5
The use of a control baseline eliminates the need for paired normal-tumour samples required in other NGS-based MSI analyses.17 Our method builds on the mSINGS tool18 using the frequencies of allele lengths different from wildtype allele in contrast to the absolute number of repeat lengths in control baseline, allowing accurate detection of low-level MSI in normal tissues indicating CMMRD. Recently, another NGS-based approach has been developed for MSI detection in blood samples of patients with CMMRD.13 A good correlation of MSI scores between both approaches was seen in shared samples provided by the C4CMMRD consortium (R2=0.91; online supplementary figure S7), suggesting that NGS-based hs-MSI assays can reliably detect CMMRD. Interestingly, our MSI score did not overlap between CMMRD samples and controls even in aplastic samples.13 The improved separation is likely due to the higher number of microsatellite markers analysed, although marker selection, bioinformatics pipeline and analysis method might also be involved.
The high accuracy and suitable turnaround time of the hs-MSI approach, similar to the recently reported in vitro MMR assay in lymphocytes,6 makes it a valuable CMMRD diagnostic tool. Since CMMRD can present with non-malignant features that overlap with NF1 and Legius syndrome, our approach could be used as a CMMRD indicator in healthy children under suspicion without germline mutations in NF1 or SPRED1, prior to MMR genes analysis, avoiding the potential pitfalls linked to the diagnosis of LS in a minor or VUS identification.7
The detection of MMR deficiency or elevated MSI score in lymphocytes may suggest pathogenicity of identified germline MMR variants (online supplementary table S7). However, caution should be taken since other variants in cis (not yet identified) could be responsible of the phenotype. The most intriguing variant in our series is MLH1 c.2146G>A (p.Val716Met). The presence of an additional causative variant on this MLH1 allele in patient E was excluded by transcript analysis.13 Although it was classified as neutral by multifactorial analysis, its identification in trans with a pathogenic MLH1 mutation in another individual with CMMRD clinical features,19 and its slightly decreased expression and MMR activity observed in heterologous systems (http://www.insight-database.org/classifications), suggest that its classification should be revisited, particularly since a hypomorphic nature cannot be totally excluded.
Mutations in MSH2 and MLH1 are associated with a more severe phenotype than MSH6 and PMS2 mutations in LS,1 and this may hold true also in CMMRD,3 although phenotype/genotype correlation in the latter is complicated by its low prevalence and the presence of hypomorphic MMR mutations.5 Even though MSI in MSH6 carriers is more precisely assessed in mononucleotide than dinucleotide repeats, higher instability levels were detected in MSH2 biallelic carriers than MSH6 carriers in our hs-MSI approach. Although the limited sample size precludes any conclusion, the MSI level may reflect the intrinsic MSH6 protein repair capacity of the particular type of markers included in the panel or could be related to disease expressivity. In contrast, no apparent differences by affected gene were observed in CMMRD lymphocytes’ MMR assay,6 which assesses the repair of a 3’-nicked G-T mismatch. Interestingly, with this method intermediate results of MMR activity and complementation were identified in some individuals, suggesting variant hypomorphic nature.6 The analysis of hs-MSI in these cases would be of particular interest.
In contrast to the absence of significant instability seen in LS samples using the hs-MSI approach, previous works described low-level MSI in blood samples by small-pool PCR15 and clone analysis.14 Although those markers were included in our custom panel design, none of them could be analysed due to insufficient coverage, with the exception of D2S123, which did not show instability in LS samples (online supplementary table S5). To improve the sensitivity of MSI assessment, the use of probes with double unique molecular identifiers tagging double-strands20 may potentially reduce the error rate and increase the sensitivity in MSI detection.
In conclusion, the high performance of the hs-MSI approach in detecting MSI in non-neoplastic tissue from patients with CMMRD is a valuable diagnostic tool which has potential in pretest selection of healthy paediatric patients, as well as in discrimination between CMMRD and other clinically related syndromes. Further evaluation in larger prospective series, including other target tissues and different disease progression stages, is needed to validate the hs-MSI approach in CMMRD diagnostic routine.
Acknowledgments
We thank the participating patients and families.
References
Footnotes
GC and MP are joint senior authors.
MG-A, FM and BP are joint first authors.
GC and MP contributed equally.
MG-A, FM and BP contributed equally.
Contributors MG-A, FM and BP designed and performed the research, analysed the data and wrote the manuscript. NB assisted in bioinformatics analyses. AF assisted in molecular analyses. MaN, HS, FB, SI, AV, EGG, VM, LIG-G, PG-G, BF, CK, TiR, ThR, DJ-L, AAA, IR, MiN, H-JP, SL, MS, KD, TI, UD and JB provided samples and clinical data. VM also contributed to the statistical analysis. RA assisted in molecular analyses. CL, DR and KW provided samples, supervised the study and wrote the manuscript. MP and GC conceived the project, supervised the study, analysed the data and wrote the manuscript. All authors revised and approved the manuscript. MP and GC shared the last authorship.
Funding This work was funded by the Spanish Ministry of Economy and Competitiveness and cofunded by FEDER funds - a way to build Europe (grant SAF2015-68016-R), CIBERONC and the Government of Catalonia (grants 2017SGR1282 and PERIS SLT002/16/0037), and the AECC fellowship to MG-A. AF was supported by a grant from the Catalonian Health Department (SLT002/16/00409). FM was supported by CIBERONC. We thank the CERCA Programme for institutional support.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval The study was approved by the ethics committee of the Bellvitge University Hospital (HUB) (file no PR255/15). Written informed consent was obtained from all individuals.
Provenance and peer review Not commissioned; externally peer reviewed.