Article Text

Abstract
Background The 15q11.2 deletion is frequently identified in the neurodevelopmental clinic. Case–control studies have associated the 15q11.2 deletion with neurodevelopmental disorders, and clinical case series have attempted to delineate a microdeletion syndrome with considerable phenotypic variability. The literature on this deletion is extensive and confusing, which is a challenge for genetic counselling. The aim of this study was to estimate the effect size of the 15q11.2 deletion and quantify its contribution to neurodevelopmental disorders.
Methods We performed meta-analyses on new and previously published case–control studies and used statistical models trained in unselected populations with cognitive assessments. We used new (n=241) and previously published (n=150) data from a clinically referred group of deletion carriers. 15q11.2 duplications (new n=179 and previously published n=35) were used as a neutral control variant.
Results The deletion decreases IQ by 4.3 points. The estimated ORs and respective frequencies in deletion carriers for intellectual disabilities, schizophrenia and epilepsy are 1.7 (3.4%), 1.5 (2%) and 3.1 (2.1%), respectively. There is no increased risk for heart malformations and autism. In the clinically referred group, the frequency and nature of symptoms in deletions are not different from those observed in carriers of the 15q11.2 duplication suggesting that most of the reported symptoms are due to ascertainment bias.
Conclusions We recommend that the deletion should be classified as ‘pathogenic of mild effect size’. Since it explains only a small proportion of the phenotypic variance in carriers, it is not worth discussing in the developmental clinic or in a prenatal setting.
- 15q11.2 copy-number variants
- neurodevelopmental disorders
- epilepsy
- congenital heart disease
- loss-of-function intolerance
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- 15q11.2 copy-number variants
- neurodevelopmental disorders
- epilepsy
- congenital heart disease
- loss-of-function intolerance
Introduction
Translating the results of clinical series and case–control studies into clinical practice is a difficult task especially for genetic variants with moderate or small effect sizes and incomplete penetrance.1 The 15q11.2 deletion located between breakpoints 1 and 2 (BP1–BP2) (MIM:615656) has been associated with neurodevelopmental disorders (NDDs), and a mild enrichment of the deletion is observed in individuals with schizophrenia (OR 1.8),2 developmental delay (OR 2.36),3 epilepsy (OR 4.9)4 and learning disabilities (OR 4.4, dyslexia and dyscalculia combined).5 The deletion has also been associated with congenital heart disease (CHD).6–8 More than 200 15q11.2 deletion carriers have been reported in clinical series with mild, moderate and severe neurodevelopmental symptoms as well as malformations leading authors to delineate a microdeletion syndrome with considerable phenotypic variability9 and incomplete penetrance.10
The BP1–BP2 region span approximately 500 kb and contains four evolutionarily conserved and non-imprinted genes (NIPA1 (MIM:608145), NIPA2 (MIM:608146), CYFIP1 (MIM:606322) and TUBGCP5 (MIM:608147)).11 These four genes are expressed in the central nervous system. Several studies have demonstrated that altered CYFIP1 expression impact neuronal function and morphology.12 13 Although CYFIP1 intragenic mutations have not yet been associated with neurodevelopmental disorder, it is considered as a primary candidate gene for the NDDs observed in 15q11.2 deletion carrier patients. Heterozygous dominant negative mutations in NIPA1 are associated with autosomal dominant hereditary spastic paraplegia type 6.14
The literature on the 15q11.2 deletion is extensive but confusing, with many studies focused on characterising carriers referred through neurodevelopmental clinics. Since most clinicians rely on descriptions from clinically referred samples, the interpretation and the counselling remains challenging for this variant frequently identified in the clinic.15
The overarching aim of the paper is to provide an accurate estimate of the effect size on cognition and risk for neurodevelopmental symptoms conferred by the 15q11.2 deletion. Specifically, we (1) investigated the association between the 15q11.2 deletion and NDDs as well as IQ, (2) investigated the association between 15q11.2 deletion and specific developmental diagnoses and (3) investigated the effect on neurodevelopmental conditions of deletions with similar probability to be loss-of-function intolerance (pLI) score (table 1).
Study aims and corresponding methods
Supplemental material
To this mean, we used new and previously published data. To accurately estimate the frequency of the 15q11.2 CNVs, analyses were computed using the largest control groups from unselected populations. To understand the discordance with frequencies and risks classically provided during genetic counselling, we compared our estimates to observations in a large clinical series of deletions (n=391) clinically referred for a neurodevelopmental disorder. To characterise the bias introduced by clinical referral, we use a control neutral CNV (15q11.2 duplication). Our estimates of the frequencies for medical and psychiatric issues in 15q11.2 deletion carriers are much lower than what has been previously reported and have implications on the relevance of reporting this CNV back to patients and families.
METHODS
Data sets
Cohorts of patients with a neurodevelopmental disorder referred for CMA analysis (aim 1)
We used data from the chromosomal microarray (CMA) diagnostic databases of the cytogenetic laboratory at Saint-Justine University Hospital (SJCHU) and Odense University Hospital (OUH) including 14 463 and 985 probands referred for NDDs and/or malformations for our enrichment analyses of the 15q11.2 CNVs.
Unselected population used as a control group (aims 1 and 2)
We used data on CNV frequency from the UK BIOBANK (151 619 individuals),16 which is an unselected population. This control data were used to compute the enrichment in NDDs (aim 1, table 2) and the meta-analyses on the association with specific diagnoses (aim 2, figures 1 and 2).
Enrichment of the 15q11.2 deletion in two independent cohorts of patients referred for neurodevelopmental disorders

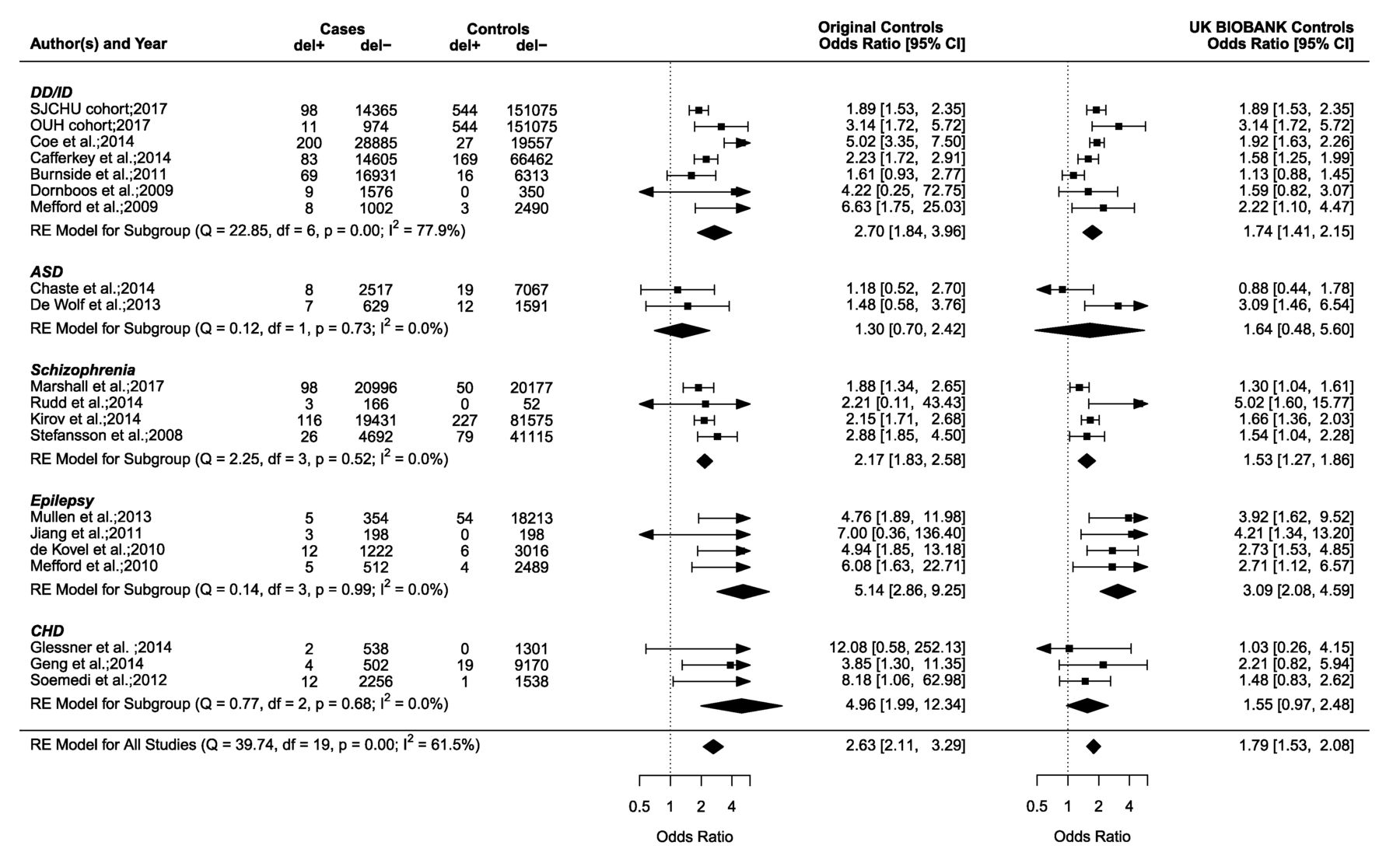
The 15q11.2 deletion case–control studies of neurodevelopmental disorders and CHD. Forest plot showing the results of 20 studies2 4 6–8 15 18–29 including the SJCHU and OUH cohorts examining the association between the 15q11.2 deletion and the OR of a neurodevelopmental disorder or CHD in cases versus controls. Of note, the year reported for the SJCHU and OUH cohorts refers to the year the data were extracted and analysed. Previously published studies are detailed in the online supplementary table S1. If a study contains a value of zero, we added 0.5 as suggested by the rma function of the metafor package. Data in the individual studies are reported with corresponding 95% CI based on a RE model. The horizontal whiskers correspond to the 95% CI for each study. The sizes of the box areas are proportional to the weight of the individual study in the meta-analysis. The width of the diamond is a summary estimate of the CI, and the dotted vertical line shows the null value of the OR and is equivalent to no effect. OR in the left side shows the OR and 95% CI for studies including the original control population, and OR in the right side shows the OR and 95% CI for the recomputed studies with the UK BIOBANK as control population (n=151 619). The test for heterogeneity (Q=39.74, df=19, p=0.00, I2=61.5%) for OR (left side) using the original control population suggests important heterogeneity among the studies. The I2 statistic describes the percentage (0%–100%) of total variation across studies that is due to heterogeneity rather than chance. The 0% indicate no observed heterogeneity, while larger values show increasing heterogeneity.36 Publication bias was assessed in a funnel plot (online supplementary figure S2), and the Egger’s test was used to test for funnel plot asymmetry (z=2.1337, p=0.0329). A p value below 0.05 suggests the presence of publication bias in the funnel plot. ASD, autism spectrum disorders; CHD, congenital heart disease; del+, cases or controls with the 15q11.2 deletion, del−, cases or controls without the 15q11.2 deletion; DD/ID, developmental delay/intellectual disability; df, number of studies; OUH, Odense University Hospital; Q, χ2 test of heterogeneity; RE, random effect; SJCHU, Saint-Justine University Hospital; SZ, schizophrenia.

{kind=link}
{kind=link}
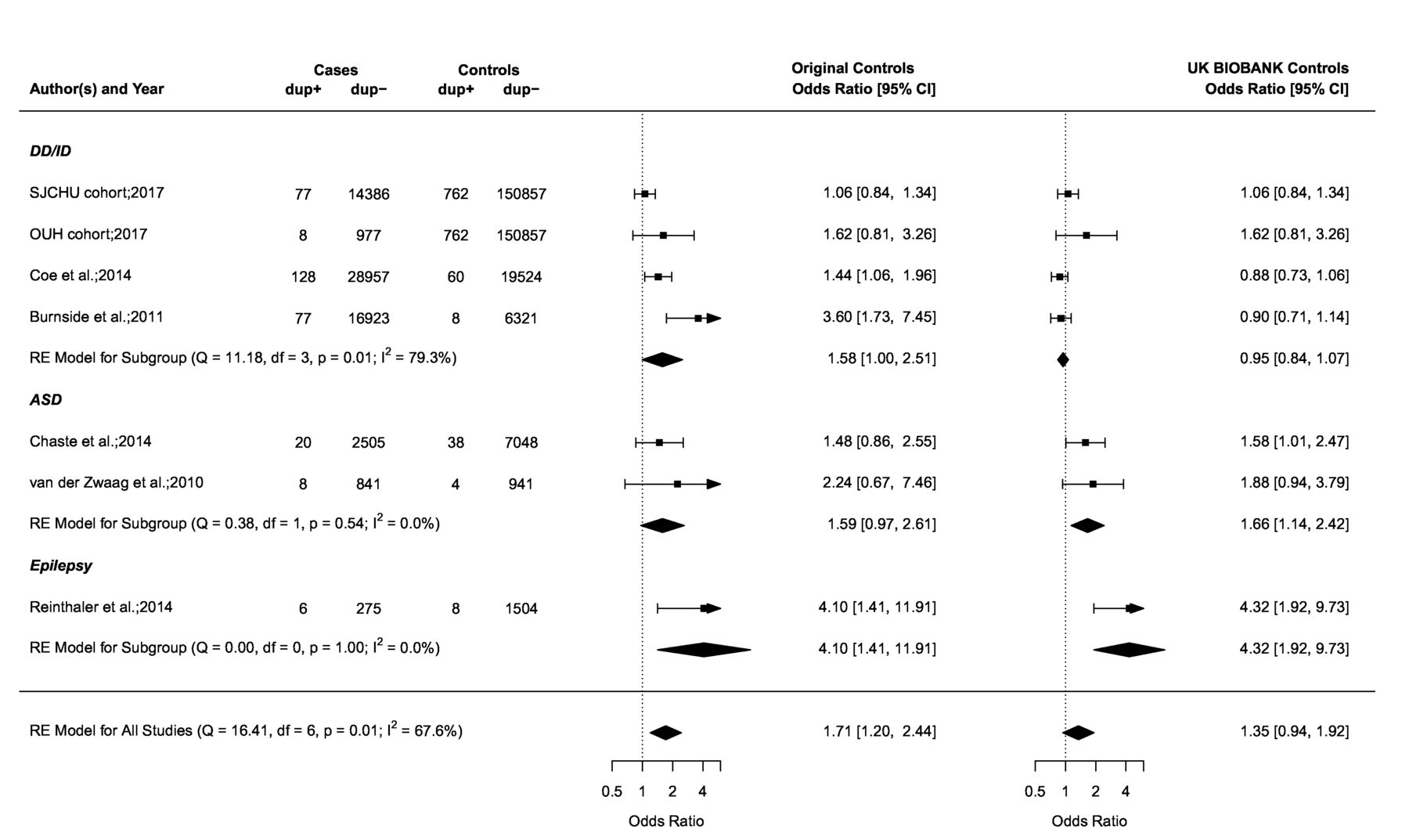
The 15q11.2 duplication case–control studies of neurodevelopmental disorders. Forest plot showing the results of seven studies18 20 21 30 31 including the SJCHU and OUH cohorts examining the association between the 15q11.2 duplication and the OR of a neurodevelopmental disorder in cases versus controls. Of note, the year reported for the SJCHU and OUH cohorts refers to the year the data were extracted and analysed. Published studies are detailed in the online supplementary table S2. Data in the individual studies are reported with corresponding 95% CI based on a RE model. The test for heterogeneity (Q=16.41, df=6, p=0.01, I2=67.6%) for OR (left side) using the original control population suggests important heterogeneity among the studies. Publication bias was assessed in a funnel plot (online supplementary figure S3) and the Egger’s test was used to test for funnel plot asymmetry (z=3.3826, p=0.0007). This result suggests asymmetry in the funnel plot, and the presence of publication bias. ASD, autism spectrum disorders; dup+, cases or controls with the duplication; dup−, cases or controls without the duplication; DD/ID, developmental delay/intellectual disability; df, number of studies; OUH, Odense University Hospital; Q, X2 test of heterogeneity; RE, random effect; SJCHU, Saint-Justine University Hospital. See figure 1 legend for more details.
DECIPHER data set used to compute de novo frequency (aim 1) and frequencies of developmental conditions in carriers of CNVs with a pLI between 1 and 3 (aim 3)
A data set including all 28 970 cases from the Database of Chromosomal Imbalance and Phenotype in Humans using Ensemble Resources (DECIPHER)17 (https://decipher.sanger.ac.uk) was received and accessed March 2018. Phenotypic information concerning CHD and epilepsy as well as information about transmission were extracted and evaluated for patients who carry either a BP1–BP2 15q11.2 CNV (n=429), or a CNV (n=382) with an annotated pLI score between 1 and 3 (described below).
Data sets for the meta-analysis of autism, schizophrenia, epilepsy and CHD (aim 2)
We performed a PubMed search and included all studies reporting the 15q11.2 CNVs between 2007 and 2017. We also searched the reference lists of retrieved studies and reviews to identify additional studies of relevance. For the meta-analysis shown in figures 1 and 2 using case–control studies, we identified 24 publications on the 15q11.2 deletion and seven on the duplication. We excluded six deletion and two duplication studies due to overlap between cases reported. Our final analysis included 20 studies2 4 6–8 15 18–29 on the deletion and seven18 20 21 30 31 on the duplication including the SJCHU and OUH cohorts (figures 1 and 2 and online supplementary table S1 and S2).
15q11.2 CNV carrier groups (clinically referred) (aim 2)
We compared our estimates of frequencies for neurodevelopmental symptoms and diagnoses computed in aims 1 and 2 to observations from a large clinical series incorporating data from 391 deletion carriers (326 probands and 65 related carriers) and 214 duplication carriers (176 probands and 38 related carriers) (table 3). Data were acquired from three different sites: SJCHU, Canada (n=220), CHUV Lausanne, Switzerland (n=143) and OUH, Denmark (n=28). We further included previously published data from 185 (150 deletions and 35 duplications) carriers.
Gender, age and inheritance status by carrier group and ascertainment
Inclusion criteria
Inclusion criteria include the presence of the recurrent 15q11.2 deletion or duplication between BP1 and BP2 (22.8–23.0 Mb, according to the human genome build CRCh37/hg19). Carrier relatives were defined as relatives who carry the same CNV as the proband. 15q11.2 deletion and duplication carriers with additional pathogenic CNVs, single-nucleotide variants (SNVs) or structural chromosomal variants were not excluded from the analyses as our working hypothesis is that every proband with a 15q11.2 deletion or duplication carry additional pathogenic variants.
Information about the participants was gathered by completion of questionnaires by the referring clinician/review of patient files (n=363) or by full assessment (n=28). We also included participants from Unique the chromosomal disorder support group, UK (n=16), and the US based 15q11.2 duplication network (Facebook group) (n=13). These participants completed self-administered questionnaires and/or provided copies of hospital files and reports. Participant ascertainment is detailed in the online supplementary materials and methods S1.1.
Among 322 deletion probands with available data, 88.8% (n=286) were children (<18 years) and 11.2% (n=36) were adults (≥18 years) at the time of ascertainment. Frequencies were similar in 170 duplication probands with 86.4% (n=152) being children and 10.2% (n=18) adults. Most deletion 79.7% (n=51) and duplication 79.4% (n=27) carrier relatives were adults (table 3).
Cognitive, psychiatric and behavioural assessments in the 15q11.2 CNV carrier groups
We collected information about global cognitive functioning for all carriers from the clinical referred group who had been assessed by different standardised neuropsychological tests as part of their initial diagnostic evaluation. The different scales used to obtain Full Scale Intelligence Quotient (FSIQ) and results are detailed in online supplementary materials and methods S1.2 and online supplementary table S3. A FSIQ of 70 or below was considered diagnostic for intellectual disability (ID). IQ range 55–70 was defined as mild ID, IQ range 40–55 as moderate and severe as IQ<40. For carriers without available FSIQ score, but with information about the spectrum of ID severity, global developmental delay, a history of learning difficulties or unspecified cognitive deficits, we calculated the frequency after re-grouping the information into the following ID categories; unspecified, mild, moderate, severe or profound (online supplementary table S3). Additional diagnosis defined by the Diagnostic and Statistical Manual of Mental Disorders, fifth edition criteria32 diagnosed in carriers before inclusion in the study are also reported in the online supplementary table S3.
Microarray platforms and additional pathogenic variants in the 15q11.2 CNV carrier groups
The majority of participant samples had been analysed across nine custom chromosomal microarray platforms (online supplementary materials and methods S1.3). All gene annotations of CNVs in this paper refers to the human genome build GRCh37/hg19. 40 (27 deletion and 13 duplication) probands carry additional pathogenic CNV (online supplementary table S4). These CNVs and the guidelines used for the interpretation are detailed in online supplementary table S5 and supplementary materials and methods S1.4. Another 10 probands (7 deletion and three duplication) were known to carry additional pathogenic or likely pathogenic (n=1) SNVs or structural chromosomal rearrangements (online supplementary table S6).
Written informed consent was obtained from patients, parents or guardian when appropriate.
Analyses
Case–control enrichment analysis (aims 1 and 2)
In order to characterise the potential unique phenotype(s) of the 15q11.2 deletion, we compared all analyses with the reciprocal duplication considered to represent a neutral CNV. Fisher’s exact test was used to assess the association between binary variables. Significance levels (p value), ORs and 95% CI were calculated using a two-tailed Fisher’s exact test. Statistical significance was set by a p value <0.05. All statistical analysis was conducted in R (V.3.3.3, the R Project for Statistical Computing; http://www.R-project.org/).
Estimating effect size on IQ using OR for ID (aim 1)
The conversion of ORs into IQ shift of CNV enrichment in cohorts of patients with NDDs is based on a model adapted from Huguet and colleagues (2018) detailed in the online supplementary materials and methods S1.5.33
Logistic regression to estimate the de novo occurrence of deletions (aim 1)
The logistic regression was performed using ‘lrm’ function from the ‘rms v5.1–2’ package34 (online supplementary figure S1 and supplemental materials and methods S1.6).
Meta-analyses (aim 2)
We used the metaphor package35 of the R Statistical Package to perform the meta-analyses in figures 1 and 2. The package has functions for fitting both fixed-effects and random-effects meta-analytic models. We fitted a random-effects model to the data used. We assessed statistical heterogeneity between studies with the Q (χ2) test and I2 statistic (total heterogeneity/total variability).36 Heterogeneity was considered significant when p<0.05. Publication bias was evaluated with the contour-enhanced funnel plot with reference line in zero (online supplementary figures S2 and S3). Funnel plot asymmetry was evaluated with a regression test (Egger’s test).
OR, relative risk (RR) and frequency (aim 2)
ORs were computed for the enrichment of the 15q11.2 deletion in autism, epilepsy and CHD. RRs of the presence of a symptom in deletion carriers compared with non-deletion carriers were computed for table 4. Of note, OR and RRs are very similar due to the low frequency of the CNVs. Frequencies (or penetrance) of a symptom or diagnosis in CNV carriers were computed by multiplying the RR by the frequency of the symptoms in the general population (table 4).
Frequencies of psychiatric and medical issues in 15q11.2 CNV carriers estimated based on case–control studies and reported in clinical series
Investigating CNVs with similar pLI (aim 3)
Genes are annotated using the pLI, where 1 means that the gene is intolerant.37 The default value associated to a gene without available score was 0. The score attributed to a CNV is the sum of scores of all genes deleted by the CNV (online supplementary materials and methods S1.7).
Additional pathogenic variants present in deletion and duplication carriers
We compared the frequency, median size (Wilcoxon Rank Sum test), mean number of genes (Student’s t-test) and the mean pLI score (Student’s t-test) of additional CNVs between deletion and duplication probands (online supplementary figures S5A-C).
Results
Enrichment of 15q11.2 deletions and duplications in clinical cohorts
The deletion is significantly enriched in the SJCHU (OR 1.9, 95% CI 1.5 to 2.4) and OUH CMA database (OR 3.1, 95% CI 1.6 to 5.7), compared with the deletion prevalence from the UK BIOBANK (0.36%, 544/151619)16 (table 2). The enrichment is consistent with our meta-analyses of previously published studies in NDD cohorts (OR 1.7–2.7, figure 1). The reciprocal duplication is not enriched in both the SJCHU and OUH cohorts (table 2) when using the prevalence in an unselected population provided by the UK BIOBANK (0.5%, 762/151619)16 and is therefore used as a control neutral CNV throughout the rest of this study.
The enrichment of the deletion in clinical cohorts are likely driven by the main referral motifs in probands, which are neurodevelopmental symptoms, epilepsy and CHD. We therefore investigated the association of the deletion with those symptoms and used the reciprocal duplication as a control.
Effect on IQ
Neurodevelopmental symptoms, and in particular cognitive deficits, represent a major referral motif for clinical cohorts. Using our model33 based on observation of IQ measured in deletion carriers in the general population, we estimate that the 15q11.2 deletion decreases IQ by 4.3 points (95% CI 6.5 to 2.03). Assuming an additive model, this effect translates into a 4% risk for ID (compared with 2% in the general population).
This mild effect is in strong contrast with the ID frequency of 42.3% (138/326) reported in deletion-carrying probands from the clinically referred group (table 4). In the same group, the mean full scale IQ (FSIQ) (available in 20/138) of deletion-carrying probands is 70.9 (±15.3 SD) (online supplementary table S3). The level of bias of the clinically referred group is illustrated by the duplication, which is not enriched in clinical cohorts and should therefore not have any detectable effect on IQ. ID is reported in 35.2% (62/176; 95% CI 28.2 to 42.8) of duplication-carrying probands (table 4) and their mean FSIQ is 75.3 (±23.0 SD) (online supplementary table S3).
Our previous study established a tight relationship between de novo frequency of CNVs and their effect size on IQ.33 The de novo frequency observed in the deletion carriers from the clinically referred group is 5.3% (8/151; 95% CI 2.3 to 10.2) (table 3 and online supplementary table S7). Merging the data from the clinically referred group and the DECIPHER data set (n=306) (online supplementary table S8) provides a de novo frequency of 8.3% (38/457; 95% CI 6.0 to 11.2). The estimated de novo frequency for any deletion with a pLI of 1.69 is 7.5% (95% CI 6.3 to 8.9, p=2e−71) (online supplementary figure S1) and is consistent with an impact on IQ of 4.3 (95% CI 6.5 to 2.03) points.
Autism and other psychiatric disorders
Autism is also a major referral motif for genetic testing. Our meta-analysis of case–control studies (figure 1) did not show a significant association between the deletion and autism spectrum disorder (ASD) (OR 1.30), which again is discordant with the frequency of ASD in deletion (12.9%, 42/326)) and duplication-carrying probands (13.6%, 24/176)) from the clinically referred group (table 4 and online supplementary table S9).
Although schizophrenia is not a referral motif for the neurodevelopmental clinics, we nevertheless performed a meta-analysis, which shows the same OR whether we used controls provided by the initial studies or UK BIOBANK data. This translates into a 2% risk for schizophrenia based on a population prevalence of approximately 1.0% for this condition.38
Epilepsy and neurological symptoms
Our meta-analysis of case–control studies for epilepsy in 15q11.2 deletion carriers4 23 26 27 shows an OR of 5.14 when using the small control groups from the original studies and 3.09 using the UK BIOBANK data (figure 1). Based on the prevalence of epilepsy in the paediatric population of 6.8 per 1000 children,39 we estimate the frequency of epilepsy in deletion carriers at 3.5% or 2.1% (table 4). This is in strong contrast with the frequency of epilepsy reported in deletion-carrying probands (15.3%, 50/326)) from the clinically referred group (table 4). A similar frequency of epilepsy is observed in carriers of the neutral duplication (14.2%, 25/176)) (table 4 and online supplementary table S10). A broad spectrum ranging from (benign) focal epilepsies to severe syndromic epilepsies is reported in deletion and duplication-carrying probands, with a predominance of generalised seizures in both deletion (6.4%, 21/326)) and duplication probands (5.7%, 10/76)). None of the relatives who carry a 15q11.2 CNV reported epilepsy (online supplementary table S11 and S12). A wide range of additional neurological manifestations is reported in deletion and duplication carriers at similar frequencies (online supplementary results S1.1 and S1.2 and online supplementary table S11 and S12).
Congenital heart disease
Our meta-analysis of CHD case–control studies6–8 shows an OR of 4.96 when the small control groups from the original publications are used (figure 1). There is, however, no significant enrichment 1.55 (95% CI 0.97 to 2.48) if the UK BIOBANK population frequency of 0.36% is used as the control group. This suggests that the frequency of CHD in deletion carriers is 4.5%–1.4% based on the prevalence of CHD of 9 per 1000 live births (table 4).40 Again the frequency reported in deletion-carrying probands from the clinically referred group is high (8.9%, 29/326) (table 4) but it is not significantly different from the frequency of CHD in duplication-carrying proband from the clinically referred group (5.7%, 10/176; 95% CI 2.8 to 10.2) (online supplementary table S13). In the same group, there were no other recurrent major malformations, medical conditions or dysmorphism reported in carriers of the 15q11.2 CNVs (online supplementary results S1.3, S1.4 and S1.5 and online supplementary table S14 and S15).
Additional pathogenic variants in 15q11.2 CNV probands
The frequency of additional CNVs reported as pathogenic, in the clinically referred group is similar in probands carrying a deletion (8.3%, 27/326) and duplication (7.4%, 13/176). The mean number of genes, the mean pLI score and the median size of additional pathogenic autosomal CNVs are also similar in deletion and duplication proband carriers (online supplementary figures S4A-C). Removing deletion and duplication probands with additional pathogenic CNVs, SNVs and structural chromosomal rearrangements from all analyses detailed above did not change any of the results (online supplementary table S9, S10, S13 and S16).
The general effects of deletions with pLI scores similar to the 15q11.2 deletion
Our previous study suggests that the sum of pLI of genes included in a deletion is tightly correlated to its effect size on cognition.33 We investigated features of deletions with a pLI sum similar to the 15q11.2 deletion (pLI sum=1.69). In the SJCHU database, the diagnostic classification of 406 CNVs with a pLI between 0.5 and 3 is benign for 60.6% (n=246), unknown for 7.6% (n=31) and pathogenic for 31.8% (n=129) (online supplementary figure S5).
The frequency of epilepsy in 442 deletions with a pLI score between 1 and 3 from the DECIPHER (n=382) and SJCHU (n=60) database is 9.3% (30/321, 95% CI 6.4 to 13.1). The frequency of CHD in deletions with 1<pLI <3 in the DECIPHER and SJCHU database is 2.5% (8/321, 95% CI 1.1 to 4.9).
Discussion
Our study provides estimates for the effect size of the 15q11.2 deletion on cognition as well as the risk of medical, behavioural and cognitive symptoms in deletion carriers. We use statistical models trained in the general population and performed meta-analyses of case–control studies using disease cohorts and control subjects. The effect size of the 15q11.2 on general intelligence is very mild with a decrease of 4.3 points in IQ, which is equivalent to a shift in IQ of 0.28 z-scores. This is identical to the estimate of the impact on IQ measured in carriers of the 15q11.2 deletion in the general Icelandic population41 and translates into a frequency of ID of 4.0% (compared with 2% in the general population). This is in line with the enrichment of the deletions in neurodevelopmental cohorts (OR 1.7, figure 1).
Our estimates for the frequency or ‘penetrance’ of schizophrenia, epilepsy and heart malformation in deletion carriers are 2.0%, 3.5% and 4.5%, respectively. These estimates could be as low as 1.5%, 2.1% and 1.4%, respectively, if one uses the population frequency of the deletion estimated in the UK BIOBANK (table 4). In the latter case, there is no significant enrichment detected for schizophrenia and CHD. In all cases, we did not detect any increased risk for ASD. Many initial case–control studies overestimated the enrichment of the 15q11.2 deletion in clinical cohorts due to small control groups providing incorrect estimates of the deletion frequency in the general population. Currently, the best estimates range from 0.36% (UK BIOBANK)16 to 0.24% (Icelandic population).41
The effects of the 15q11.2 deletion may point towards important biological mechanism, but they are too small to support any relevant counselling in the clinic. The deletion should not be considered as an etiological factor for patients referred to the developmental clinic. In particular, the SD of IQ observed among siblings (±12 points around the parental mean)42 is three times higher than the effect size of the 15q11.2 deletion. The presence or absence of the deletion in a young child is therefore a very poor predictor of developmental delay or IDs. Our estimate of the effect size on IQ is concordant with the frequency of de novo occurrence observed in deletion carriers (8.3%), which is tightly related to the effect size of deletions on IQ.33
Clinical series have been useful to delineate rare syndromes with remarkable and/or severe presentations. Using the duplication as a ‘neutral CNV’ control group, we show that the average proband referred for CMA testing presents with ID, epilepsy, autism and cardiac malformations in approximately 35%, 14%, 17% and 6% of the cases, respectively. This suggests that clinical series will report similar frequencies regardless of any formal association (ie, association of the 15q11.2 deletion with ASD and CHD has yet to be demonstrated). Clinical series are especially problematic for genetic variants with an effect size much smaller than the average referral criteria (ie, 16p13.11, 1q21.1 TAR, 17q12, 16p11.2 Distal (SH2B1), 1q21.1 Distal (Class I) and 15q13.3).33
In our clinically referred groups, we observed no significant difference in symptom frequency between deletions and duplications. Even the distribution of epilepsy types was similar in deletion and duplication carriers (generalised seizures in 6.4%–5.7%).
Based on our estimates, ID in 15q11.2 deletion carriers is mostly due to additional factors. Previous studies have suggested that the deletion may interact with other factors or that expression levels of genes within the 15q11.2 locus may underlie the phenotypic variability but a properly powered study has yet to demonstrate this.43 Instead, it is likely that the deletion acts additively on cognition with other genetic and environmental factors, which is consistent with the fact that most of the genetic contribution to general intelligence is thought to be additive.44 In this context, the contribution of the deletion to the important decrease in IQ observed in a proband carrier referred for ID would be negligible. Our investigation of non-overlapping deletions with similar effect size on cognition (based on the sum of pLI scores) show that a large proportion of these rare CNVs are classified as benign (online supplementary figure S5). If the 15q11.2 deletion had been less frequent, it would obviously not have been identified as a neurodevelopmental risk factor.
The guidelines of the American College of Medical Genetics45 46 recommend three categories of clinical significance including pathogenic, uncertain clinical significance and benign. Variants with uncertain clinical significance are subcategorised in likely pathogenic, likely benign and no subclassification. The 15q11.2 deletion has been assigned in laboratories as a variant of uncertain clinical significance; likely pathogenic as well as pathogenic with variable expressivity and incomplete penetrance. We propose that variants should be reported in medical diagnostics, when possible, with a quantitative estimate of effect size. There is strong evidence associating the 15q11.2 deletion with NDDs and a decrease in cognitive abilities so this variant should not be classified as ‘uncertain significance’. We propose a new classification of the 15q11.2 deletion as ‘pathogenic of mild effect size’. We recommend informing patients that because these variants only explain a small proportion of phenotypic variance, they are not discussed in the developmental clinic or the prenatal setting. The clinician may therefore consider additional genetic testing based on the clinical phenotype. In the near future, such variants will likely be integrated in polygenic risk scores.
Limitations
Our estimates are derived from case–control studies and statistical models trained in unselected populations. Different unselected population cohorts may provide slightly different prevalence’s of the 15q11.2 deletion and therefore change our estimates. However, many of the early case–control studies grossly underestimated the frequency of the deletion in the general population due to very small control groups.
Our statistical model estimates small effect sizes on IQ for CYFIP1, NIPA1, NIPA2, and TUBGCP5 (-2.72, –0.59, −1.33 and 0.0 points of IQ, respectively) and could underestimate the overall effect of the deletion.33 Our individual gene estimates are, however, concordant with the absence of any de novo mutations in NIPA1, NIPA2, CYFIP1 and TUBGCP5 in the public archive of interpretations of clinically relevant variants, ClinVar accessed March 2018 (http://www.ncbi.nlm.nih.gov/clinvar).47 This may lead to reassessment of the effect size of CYPFIP1, which has been proposed as a large effect size neurodevelopmental gene.12 13 Estimates and frequencies derived from case–control association studies do not allow to clearly rank the symptoms contributing to the enrichment in disease cohorts. For example, it is unclear, which features among cognitive symptoms, seizure and heart malformations are mainly driving enrichment in neurodevelopmental cohorts.
In conclusion, we recommend that the 15q11.2 deletion should not be reported back to patients, as the contribution of this variant to symptoms of a proband referred to the neurodevelopmental clinic is modest. Pursuing diagnostic analyses is recommended in any 15q11.2 deletion carrier referred for significant neurodevelopmental symptoms. This variant cannot be used as a marker for prenatal testing because it explains only a small fraction of the phenotypic variance. Series of clinically referred patients should not be used to study mild or moderate effect-size variants unless strategies such as intrafamilial control subjects are used to mitigate ascertainment bias.1 In the future, polygenic risk scores will help estimate the cumulative impact of small effect size variants identified in patients.
Acknowledgments
We are grateful to all patients and families who participated in the study and made this work possible. We are grateful to our collaborators from the 15q11.2 Working Group for contributing patients for this study. We thank Unique-The Rare Chromosome Disorder Support Group, a UK-registered charity and the online US-based 15q11.2 duplication network group for advertising the study and for referred participants. This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was provided by the Wellcome Trust. This study has been conducted using data from the Saguenay Youth Study (SYS) resource. We also want to acknowledge OPEN, Odense Patient data Explorative Network, Odense University Hospital Odense, Denmark: www.sdu.dk/ki/open. We thank Martineau Jean Louis for his help with ANNOVAR, and Catherine Schramm for her helpful advice on statistical analysis. We thank Dr. Jacquemont’s team for fruitful discussions. We are grateful for the collaboration with the Danish Departments of Clinical Genetics for sharing their data and for granting data access to Danish Cytogenetic Central Register (DCCR). We thank Jan Hansen from DCCR for his help with data retrieval.
References
Footnotes
LBO and SJ are joint senior authors.
LBO and SJ contributed equally.
Collaborators 15q11.2 Working Group: Joris Andrieux, Angela Barnicoat, Patricia Blanchet, Sophie Blesson, Florence Niel Bütschi, Philippe M Campeau, Nora Chelloug, François-Guillaume Debray, Florence Fellmann, Alessandra Ferrarini, Richard Gibbons, Pernille Axel Gregersen, Juliane Hoyer, Ulrike Hüffmeier, Ditte Kjelgaard, Mandy Krumbiegel, Sébastien Lebon, Gaetan Lesca, Stéphanie Marignier, Sandra Mercier, Jacques Michaud, Grant Mitchell, Isabelle Mortemousque, Rikke S Møller, Mathilde Nizon, Genevieve Pierquin, Kristina Pilekær Sørensen, Sue Price, Pascal H Pujol, Vincent Ramaekers, Martine Raynaud, André Reis, Massimiliano Rossi, Pierre Sarda, Franco Stanzial, Helen Stewart, Dea Svaneby, Christian T Theil, Marianne Till, Yannis Trakadis, Dorothée Ville, Sandrine Vonwill, Andrew Wilkie, Antje Wiessner. Members of the 15q11.2 Working Group are collaborators who are listed in the supplementary file.
Contributors AEJ, LBO and SJ contributed to the concept, study design and wrote the manuscript. AEJ, ED, CM, MP, CBA, EL, LBO and SJ collected the data and analysed or interpreted the data. AEJ, CM, AVD, MP, RFK, JP, CC, DS, HL, SR, AP, DG, UK, CLC, JL, ABS, BI, CZ, JHC, MAD, AB and HH recruited patients and performed the clinical evaluation of patients. All authors revised and approved the final version. Collaborators, RSM on behalf of the members of the 15q11.2 Working Group provided patients for the study.
Funding This work was supported by grants from Odense University Hospital Free Research Fund, Grant No.15-A857 (to AEJ), the Region of Zealand and Region of Southern of Denmark Joint Research Fund, (14-001308 to AEJ), the Canadian Institute of Health Research (CIHR 159734), Ph.D. Scholarship from the Region of Southern of Denmark (to AEJ) and a PhD Scholarship from the Faculty of Health Sciences, University of Southern of Denmark (to AEJ), a Bursary Professor fellowship of the Swiss National Science Foundation (SNSF) (to SJ), a Canada Research Chair in neurodevelopmental disorders (to SJ) and a Chair from the Jeanne et Jean Louis Lévesque Foundation (to SJ).
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval This study was approved by the institutional review boards (IRB) of Sainte-Justine University Hospital, Montreal, Canada (MP-21-2016-946), Centre Hospitalier Universitaire Vaudois (CHUV) in Lausanne, Switzerland (http://www.cer-vd.ch/) and the Regional Committees on Health Research Ethics for Southern Denmark (ID-20150086).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data relevant to the study are included in the article or uploaded as supplementary information.