Article Text

Abstract
Background Preimplantation genetic testing (PGT) has already been applied in patients known to carry chromosomal structural variants to improve the clinical outcome of assisted reproduction. However, conventional molecular techniques are not capable of reliably distinguishing embryos that carry balanced inversion from those with a normal karyotype. We aim to evaluate the use of long-read sequencing in combination with haplotype linkage analysis to address this challenge.
Methods Long-read sequencing on Oxford Nanopore platform was employed to identify the precise positions of inversion break points in four patients. Comprehensive chromosomal screening and genome-wide haplotype linkage analysis were performed based on SNP microarray. The haplotypes, including the break point regions, the whole chromosomes involved in the inversion and the corresponding homologous chromosomes, were established using informative SNPs.
Results All the inversion break points were successfully identified by long-read sequencing and validated by Sanger sequencing, and on average only 13 bp differences were observed between break points inferred by long-read sequencing and Sanger sequencing. Eighteen blastocysts were biopsied and tested, in which 10 were aneuploid or unbalanced and eight were diploid with normal or balanced inversion karyotypes. Diploid embryos were transferred back to patients, the predictive results of the current methodology were consistent with fetal karyotypes of amniotic fluid or cord blood.
Conclusions Nanopore long-read sequencing is a powerful method to assay chromosomal inversions and identify exact break points. Identification of inversion break points combined with haplotype linkage analysis is an efficient strategy to distinguish embryos with normal or balanced inversion karyotypes, facilitating PGT applications.
- pathogenic structural variants
- Oxford Nanopore technology
- long-read sequencing
- genome-wide haplotype analysis
- preimplantation genetic testing
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- pathogenic structural variants
- Oxford Nanopore technology
- long-read sequencing
- genome-wide haplotype analysis
- preimplantation genetic testing
Introduction
Chromosomal inversions are common structural chromosome rearrangements that occur when one chromosome breaks at two points and reinserts in reverse, including paracentric inversion and pericentric inversion. The prevalence of chromosomal inversions is estimated to be 0.96%–1.10% in patients who have a history of recurrent miscarriages or undergo in vitro fertilisation (IVF).1 2 Although inversion carriers usually have a normal phenotype, carriers may be affected by infertility due to spermatogenetic failure, miscarriages or abnormal offspring.3 4 The genetic effect of chromosomal inversion mainly depends on the length of the inverted region, for the chance of meiotic unbalances is associated with the size of the inverted segment in proportion to the whole chromosome. The rearranged chromosomes are considered as a risk factor for reproduction if the inverted segment constitutes more than 30% of the total chromosome length and more clinical care should be paid once the inverted segment exceeds 50%.3 5 Generally, chromosomes will pair with homologous fragments during meiosis in primordial germ cells, thus forming inversion loop at meiosis I and finally generating four kinds of gametes after crossover of homologous segments in the loop. Among all the gametes, one has normal sequence and another one has the inverted sequence, while the others are unbalanced with partial monosomy or partial trisomy.6 The overall risk is estimated at 5%–10% for an inversion carrier to have an affected child with unbalanced rearrangement is.7
Chromosomal rearrangements have long been known to be a risk factor for fertility. For these couples with rearrangements, preimplantation genetic testing (PGT) has been widely used to select normal or balanced diploid embryos. Initial method of FISH has obvious technical limitations such as signal interpretation and the number of tested chromosomes.8 For the past few years, molecular platforms which detect chromosome unbalances and simultaneously screen aneuploidy have significantly enhanced success rates of pregnancy.9–11 In the meantime, SNP microarray (SNP array) gives a great performance in detecting polyploidy and UPD.12 13 Despite these advantages, current methods cannot detect inversions in embryos due to the lack of chromosomal copy-number changes, and little research has been conducted in this area due to technical limitations. Many of them will pass on the inversion to their children who may also be faced with infertility or recurrent pregnancy loss.
Long-read sequencing can generate reads that are on average 10 kb or longer, which can be aligned to repetitive sequences and span break points of structural variations (SV). More than 20 000 SVs including insertions, deletions, duplications, inversions and translocations at least 50 bp in size have been reported in each human genome, through de novo human genome assemblies from PacBio single molecular real-time (SMRT) sequencing and Oxford Nanopore sequencing.14 15 Merker et al reported that a 2184 bp deletion in PRKAR1A was identified through SMRT sequencing, which was among the first successful clinical applications in identifying pathogenic variants in patients via long-read sequencing.16 In addition, Ishiura et al used Nanopore sequencing and reported abnormal expansions of TTTCA and TTTTA repeats in intron 4 of SAMD12 gene which cause adult familial myoclonic epilepsy.17 Moreover, Nanopore sequencing identified a 7 kb deletion in G6PC gene as a pathogenic variant for glycogen storage disease type Ia, while whole-exome sequencing missed the pathogenic variant since both break points were located in Alu elements.18 In summary, long-read sequencing showed obvious advantages in detecting SVs that were located in low complexity region.
The current work aims to distinguish embryos that carry a normal karyotype from those with balanced inversion karyotypes prior to implantation, to facilitate the selection of embryos for IVF via PGT. In our study, long-read sequencing on the Oxford Nanopore platform was used for identifying inversion break points, together with comprehensive chromosomal screening (CCS) and haplotype linkage analysis. Haplotypes including break point regions, the inversion chromosome and corresponding normal homologous chromosome were established. The study was performed in a blinded fashion where laboratory techniciansdid not know the true results, and the conventional amniotic fluid or cord blood cell karyotypes were used to validate the predictive accuracy.
Materials and methods
Patients
Four patients with cytogenetically validated chromosomal inversion who undergo assisted reproduction were enrolled from January 2017 to October 2018. Patient 1 experienced two repeated miscarriages, patients 2 and 3 had the history of primary infertility. Patient 4 experienced two pregnancies with fetal anomaly, both of which proved to be unbalanced inversion karyotypes with a partial trisomy and a partial monosomy by second-trimester prenatal diagnosis. In addition, these three male patients (patients 2, 3 and 4) were severe oligozo-ospermia. The karyotypes were 46,XY,inv(1)(p13;q25), 46,XX,inv(4)(p14q27), 46,XY,inv(7)(p15q22) and 46,XY,inv(10)(q11.2q21), respectively. Ten millilitres of peripheral blood was collected from each couple and family numbers at recruitment.
IVF, blastocyst biopsy and whole-genome amplification
Standard techniques were used in IVF. Briefly, Metaphase II (MII) oocytes were produced using intracytoplasmic sperm injection, and then cultured for 5–6 days to develop to the blastocyst stage. In our centre, the criteria of grading blastocysts were according to the recommendation by Schoolcraft et al.19 Three to ten cells were biopsied from the trophectoderm cells and then placed into PCR tubes as previously described.20 Whole genomic amplification (WGA) was performed using the multiple displacement amplification method (QIAGEN, Hilden, Germany).
Long-read sequencing by Oxford Nanopore technology
Genomic DNA was extracted and then we created large insert size libraries according to the recommended protocols (Oxford Nanopore, UK). Five-microgram genomic DNA of each sample was sheared to ~5–25 kb fragments using Megaruptor (Diagenode, B06010002), size selected (10–30 kb) with a BluePippin (Sage Science, MA) were conducted. Subsequently, we prepared genomic libraries using the Ligation Sequencing 1D Kit (Oxford Nanopore, UK). Ligation was performed by mixing purified dA tailed sample, blunt/TA ligase master mix (No M0367, NEB) and tethered 1D adapter mix (SQK-LSK108) and incubated at room temperature for 10 min. Finally, adaptor-ligated DNA was cleaned up. Sequencing was performed on R9.4 flowcells using GridION X5.
Short-read sequencing on MGISEQ2000
The DNA library was prepared using the MGIEasy Library Prep Kit V1.1 (MGI Tech) and loaded to MGISEQ2000. According to the manufacturer’s instructions (V1.1 and A0, respectively), reagents from the MGISEQ-2000RS High-throughput Sequencing Set (PE150) were used for massively parallel paired-end 150 bp sequencing. Raw reads with FASTQ format were generated using zebracallV2 software. All the reads were aligned to reference genome (hg19) using BWA (0.7.17-r1188). Delly (0.7.9) and manta (1.5.0) with default parameters were used for SV calling.
Detection of inversion break points and validation by Sanger sequencing
SVs were detected with NGMLR-Sniffles pipeline.21 Briefly, long reads were aligned to hg19 reference genome by NGMLR (version 0.2.6) using the preset parameter for Nanopore technology (X-ONT) and whole-genome SVs were subsequently detected by Sniffles (version 1.0.7) (--min_support 1 --num_reads_report −1). According to the result of karyotype analysis, inversion variants in suspected region were extracted. Ribbon was used to visualise the alignment results and we manually validated possible SV calls.22 All inversions in suspected region were filtered by the following criteria. At first, the support read number of an inversion is not less than 2. Second, both 5′ end and 3′ end junctions of an inversion have support reads. Inversions meeting these criteria were kept for PCR and validation by Sanger sequencing. Region-based annotation of break points was obtained using ANNOVAR.
Primers for reference sequences and break point junction sequences of both 5′ end and 3′ end of the inversion were picked through using Primer3 software. PCR was performed according to the manufacturer’s recommended protocols. PCR products of reference sequences and break point junction sequences were subjected to agarose gel electrophoresis. Products of break point junction sequences were sequenced by Sanger sequencing. Precise break point positions were identified by mapping Sanger sequencing sequences to hg19 reference genome by BLAT23 and minimap2 (version 2.10).24
SNP array and haplotype analysis
As previously described, genome-wide SNP genotypes were generated by SNP microarray (Illumina Human Karyomap-12V1.0).25 The haplotypes were established with informative SNPs, including the two break point regions, the whole inversion chromosomes and the corresponding homologous chromosomes in the couple, reference and embryos. Either an unbalanced embryo or a carrier’s family member was used as a reference to phasing haplotype. The selection criteria for informative SNPs were that they should be homozygous in the spouse and heterozygous in the patient. In addition, these SNPs in the patient’s parents or other family members should be homozygous when they were used as references to phase haplotype. The haplotypes of the whole inversion chromosome and the homologous chromosome could indicate recombination around the break points.
Results
Controlled ovarian hyper-stimulation (COH) and CCS results
In the proposed study, the four patients with chromosomal inversions underwent five IVF cycles. Patient 4 experienced two cycles and the other patients had one cycle. The characteristics and COH results of these patients are listed in online supplementary table S1. In patients 1, 3 and 4, the inversion was inherited from one parent and was de novo in patient 2. CCS was performed in all the 18 biopsied blastocysts, in which eight were diagnosed as diploid embryos with inversion or normal karyotype, five showed unbalanced abnormalities related to inversion and five had de novo chromosomal abnormalities unrelated to inversion. These results are presented in table 1. The study designation and workflow are shown in figure 1.
Supplemental material

The study designation and workflow. (A) The genetic map of an example of the recruited family, all the genetic maps of the four families were included. (B) Flow chart of the experiment and bioinformatics pipeline, the process is universal for all patients. CCS, comprehensive chromosomal screening; SV, structural variation; QC, quality control.
Detailed results of microarray platform of the biopsied blastocysts
Nanopore sequencing identified inversion break point accurately
Low-coverage (~10× coverage) Nanopore long-read sequencing was carried out and 31.3, 28.9, 33.0 and 32.1 G bases were generated with average length of 17 748, 17 114, 13 792 and 16 577 bp for the four patients, respectively (table 2). Read length distributions were similar for all the patients (online supplementary figure S1A) and mean mapping rates exceeded 91%. The overall mapping identity for long reads was about 83.3% in all samples (online supplementary figure S1B). Details of the yield, read length and accuracy of long-read sequencing were provided in online supplementary table S2. High-confident inversions for all the four patients were found. In patient 1, an inversion that spans chr4:48457186 to chr4:117253985 was identified; eight reads supported the inversion and the read numbers for 5′ end junction and 3′ end junction were 6 and 2, respectively (figure 2A). In patient 2, an inversion that spans chr1:95878163 to chr1:193437123 was identified; 11 reads support the inversion and the read numbers for 5′ end junction and 3′ end junction were 7 and 4, respectively (figure 2B). Additionally, for patient 3, an inversion that spans chr10:45936709 to chr10:61040107 was found; eight reads supported the inversion and the read numbers for 5′ end junction and 3′ end junction were 5 and 3, respectively (figure 2C). However, the SVs in patient 4 were more complicated (figure 2D), as two high-confidence inversions were found in break point region provided by cell karyotype. Five reads supported chr7:2538292–132144231 and three reads supported chr7:2538317–132149326. All the reads supporting chr7:2538292–132144231 spanned the 5′ end junction and all the reads supporting chr7:2538317–132149326 spanned the 3′ end junction. Both the inversions were consistent with the karyotype and thus we inferred that the two inversions constructed a complex ‘Deletion-Inversion-Deletion’ (DEL-INV-DEL) SV. For all the inversions, long-read supported normal chromosomes were observed, suggesting that these inversions were heterozygous in the carriers. We also applied an orthogonal sequencing platform using MGISEQ2000 to generate ~30× sequencing coverage per individual. Integrative Genomics View was used to check the short reads and long-read alignments, three of eight break points were located in repeat region. Three of four inversions were found in the SV call set using delly and manta. The complex SVs in patient 4, DEL-INV-DEL, were missed by callers using next-generation sequencing (NGS) data (figure 2). This may be due to the complex and nested SVs and the repeat enriched region.
Supplemental material
Supplemental material
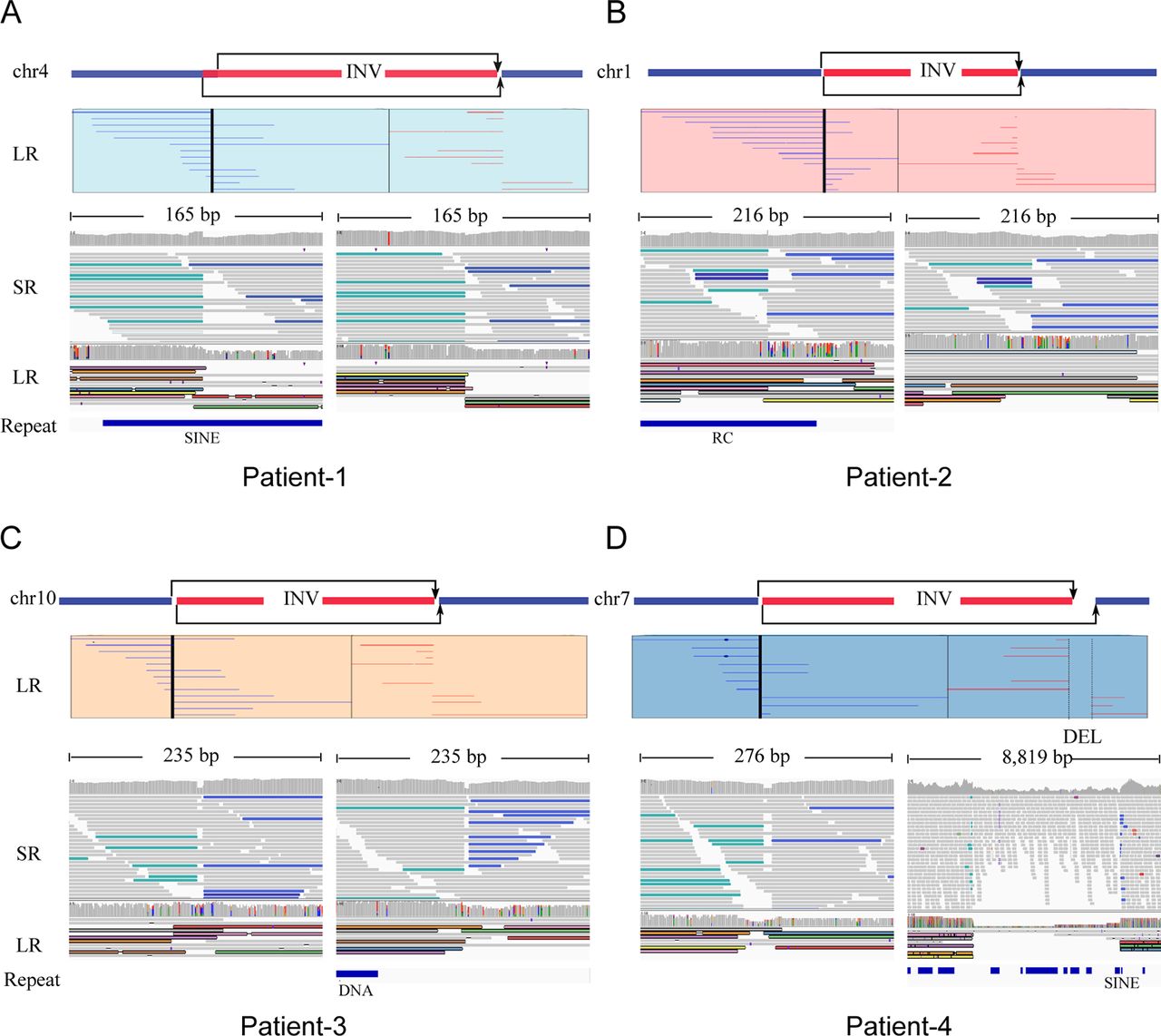
Large inversion variants detected by Nanopore long-read sequencing technology. Four cases, patients 1–4, were represented by (A), (B), (C) and (D), respectively. For each case, the top panel figured the chromosome which inversion occurred supported by karyotype results, and long-read (LR) alignments coving the break points. Two parts of the split reads were denoted by blue (forward) and red (reverse). The panel in the bottom shows short-read alignments (BWA-MEM; IGV-SR) and long-read alignments (ngmlr; IGV-LR) around break points using Integrative Genomics View (IGV). All the break points, three of eight located in repeat region, were supported by short and long reads, except one in patient 4, DEL-INV-DEL, was missed by callers by using short reads. DEL, deletion; INV, inversion; RC, Rolling Circle; SINE, Short interspersed nuclear elements.
Summary of break point characterisation results of the patients by Nanopore and Sanger sequencing
To further validate inversion calls detected by Nanopore sequencing, we performed PCR and validation by Sanger sequencing. The primer information was listed in online supplementary table S3. For each inversion, primers for 5′ end reference sequence (No 1), 3′ end reference sequence (No 2), 5′ end break point junction sequence (No 3) and 3′ end break point junction sequence (No 4) were selected. For patients 1–4, all of PCR products and break point junction sequences were observed, which confirmed the four heterozygous inversion variations. Length of PCR products was consistent with the expected product size, especially for patient 4, which inferred there was a large deletion near the break point. To further identify the precise break points, we performed Sanger sequencing on PCR products for break point junction sequences. The precise break point positions for all of the rearranged segments could be found in table 2 and figure 3B. The mean absolute distance from break point position detected by Nanopore sequencing to break point position detected by Sanger sequencing was about 12.5 bp. The sequencing results in these break point regions indicated the complexity of human genomic rearrangement.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
PCR and Sanger sequencing validated inversion variants detected by Nanopore long-read sequencing technology. (A) PCR validation of inversion variants. PCR products of the four patients and the normal control were shown in the top and bottom parts, respectively. For each individual, four fragments, two break point junctions (BJ1 and BJ2) and two normal alleles (N1 and N2), were validated. (B) Four schematic diagrams displayed precise break points identified by Sanger sequencing.
Phasing haplotype
Haplotype models were established for the carrier, the spouse and the reference, including the break point regions, the whole inversion chromosomes and the homologous chromosomes. Then, haplotype analysis was performed in the eight diploid blastocysts, suggesting that two were with normal karyotypes and the other six were with balanced inversion karyotypes. The predictive results were consistent between the break point regions in each embryo. Detailed results of the microarray platform were listed in table 1 and the summary of informative SNPs of the diploid blastocysts was shown in table 3. In patient 4, the predictive results with family members or unbalanced embryos were consistent, as shown in table 1.
Summary of informative SNPs used to establish the haplotypes of the diploid blastocysts
Clinical outcome
In patients 1 and 4, the women failed to get pregnancy in the first transfer cycle, while they were successful in the second cycle. In other patients, they got pregnant in the first cycle. Cytogenetic analysis of amniotic fluid or cord blood was required for these patients after pregnancy. We validated that the computational predictions and the cytogenetic results were totally consistent. All the newborns were healthy with normal phenotype. The sensitivity and specificity of our study were 100%, though we acknowledge that a small sample size was included in our study. In addition, the existence of the rearranged chromosomes was also predicted based on haplotype analysis. The above results further demonstrated the accuracy of the proposed approach.
Discussion
Although many patients who carry inversions can have successful IVF through PGT, many of them will pass on the inversion to the next generation who may also be subjected to infertility difficulty. Until now, no molecular method is capable of reliably distinguishing embryos that carry an inversion from those with the normal karyotype. Although in most cases there is no phenotypic difference between embryos with or without an inversion, many couples express a strong will to transfer one embryo with normal karyotype. Therefore, their offspring will not face any reproductive problems in association with this rearrangement.
Numerous researchers have been attempting to address the difficulty. For carriers with balanced translocation, the most common type, embryos with balanced translocation or normal karyotypes can be successfully identified,26 27 and the current methods required the break point identification within single base to kilobase resolution by unbalanced embryos. However, for chromosomal inversion carriers, the proportion of unbalanced embryos is low. In our centre, the proportion was 4% in all blastocysts analysed of 39 uncommon inversion patients undergoing PGT treatment. Besides, the karyotype identified by peripheral blood lymphocyte cells may lead to misjudgement of the break point region due to the limitation of low resolution of 5–10 Mb.
Break point identification is a significant problem in cytogenetics and several techniques had been developed to map chromosome break points to the kilobase level.28–30 However, these techniques are time consuming and difficult to be widely applied in clinical laboratories. Moreover, Kato et al found that rearrangement break points usually happened in complex regions, such as AT-rich short tandem repeats and long low copy repeats, and NGS methods cannot address difficulties in these cases due to technical limitations of short reads.26 31 Nanopore sequencing is an emerging technology with multiple advantages. In the current work, we successfully identified the precise break point positions of inversion variations (one inversion-ins, two inversions and one DEL-INV-DEL) with only ~10× coverage long-read sequencing data on the Oxford Nanopore platform. A recent research showed that 10–30× coverage of long reads could recover around 80% of the all types of SV calls with precision of ~80% or higher.21 However, short-read approaches have been reported to lack sensitivity (only 10%–70% of SVs detected),32 33 exhibit very high false positive rates (up to 89%)33 34 and misinterpret complex or nested SVs.21 Long reads are superior to short reads regarding detection of complex chromosome rearrangements.35 Moreover, long-read sequencing technology can sharply facilitate the discovery and genotyping of SVs, which greatly improves the ability to connect genotypes to phenotypes, showing potential important prospects of clinical application.
Based on our knowledge, this was among the first report to distinguish inversion and normal karyotypes in embryos, and among the first time that Nanopore long-read sequencing was introduced into assisted reproduction for clinical diagnosis. Several advantages could be concluded in the study. First, Nanopore long-read sequencing could identify the inversion break points and find the disrupted genes precisely. Second, the prediction for the status of chromosome inversion and the CCS in genome-wide could be performed simultaneously. Third, apart from the haplotypes of the break point regions, the haplotypes of the whole chromosome involved in the inversion and the corresponding normal homologous chromosome could be established simultaneously. Fourth, the methodology was universal for any kind of inversion theoretically. By contrast, one limitation of our research was that a small sample size was included, a prospective multicentre study with a large sample size should be performed to further confirm the feasibility. In addition, the cost of the entire experimental work is also a critical point in the PGT programmes in daily practice. The total cost of our method is not higher than common NGS strategies. The cost of identifying the break point with Nanopore sequencing in blood sample is similar to NGS, and the cost of SNP array performed in WGA production of biopsied cells is necessary in routine PGT. With the rapid development of long-read sequencing, the cost will become lower and lower. In patients 2 and 4, the unbalanced embryo was used as a reference and the predictive results with either patients’ parents or unbalanced embryos were consistent. However, compared with the patients, the unbalanced embryo could not help establish the haplotype of whole inversion chromosome as the existence of partial monosomy or trisomy of the distal regions, and therefore could not present the recombination in these regions. Moreover, we found that the frequency of homologous recombination in inversion chromosomes was significantly lower than that in the normal chromosomes, suggesting that the existence of inversion loop may reduce the chances of recombination or lead to the asynapsis between the inversion and normal chromosome, which was consistent with that reported,36 showing a detrimental effect of inversion loop on meiosis. In the current work, 7.0±0.46 SNPs/Mb could be used to establish haplotypes, the region of ±2 Mb flanking the break point was enough for phasing the haplotype.
Furthermore, among all the analysed embryos, the rate of unbalanced embryos related to inversion was 27.8% (5/18). In patients 1 and 3, no unbalanced embryos were found and the length of inverted segment might account for this. The rate of unbalanced recombinants seemed to be influenced by many factors, such as the region affected, the position of the break points, the chromosome involved or the size of the inverted segment. As reported, no unbalanced recombinants were produced when the inverted segment involved <30% of the chromosome length in spermatozoa.37 Between 30% and 50%, the inverted segment would induce a slightly increased risk of the unbalanced recombinants, and the risk became very important when the inverted segment was >50% of the chromosome length.2 37 Luo et al found the frequency of the recombinant gametes showed direct relation with the proportion of the inverted chromosome region by sperm-FISH method.38 Moreover, the embryo 1 of patient 1 was a UPD (1–22, X) which was diagnosed through CCS with SNP array. UPD is defined as the inheritance of both homologues of a chromosome from a single parent and no contribution from the other parent.39 40 This phenomenon is primarily thought to be the result of trisomy self-correction mechanism, which occurs when a trisomic cell attempts to restore disomy by excluding one of the three homologues.41 42 Liehr et al reported that there was an increased risk of UPD in the offspring of chromosome rearrangement carriers due to the complex segregation patterns.43 Therefore, it is important to note the limitations in detecting UPD. NGS and array-comparative genomic hybridization (CGH) platform could not detect either heterodisomy or isodisomy,13 whereas SNP microarray method has been reported successfully to detect UPD.12 44 Thus, for PGT platforms of array-CGH and NGS, patients with embryos that have been diagnosed as diploid should be counselled about the limitations of the detection technologies.
In conclusion, we showed that the identification of chromosomal inversion break points combined with haplotype linkage analysis is an efficient method to distinguish embryos that carry a balanced inversion from those with a normal karyotype. This study provides significant clinical implications for these patients and enables more accurate genetic counselling for patients carrying chromosomal inversions. More inversion patients can benefit from stopping passing on the rearrangement to their children. However, specificity and sensitivity of our methods should be further verified with larger sample sizes.
Acknowledgments
We express deepest gratitude to all the families enrolled in our research. We also thank the staff of the Genetics Laboratory and IVF Laboratory for their invaluable efforts and contribution relating to the experiment. We thank Dr Kai Wang (Perelman Center for Cellular and Molecular Therapeutics, Children’s Hospital of Philadelphia, USA) sincerely for providing valuable feedback on the use of bioinformatics tools to analyse long-read sequencing data and detect structural variants.
References
Footnotes
SZ and FL contributed equally.
Contributors CX, YL and XS designed the research. SZ and FL wrote the manuscript. SZ, FL, CL, JW, JF, QY, GY, YZ, XL and XS executed the research (SZ, FL and QY performed the data analysis; SZ and CL performed the microarray experiments; XL performed the PCR validation; JF performed the intracytoplasmic sperm injection and blastocyst biopsy experiments; JW, XS, CL and YZ collected the cases). GY, DW, XS, DL and CX directed the critical discussion of the manuscript. All authors approved the final manuscript.
Funding The research was supported by the National Key R&D Program of China (2016YFC1303100), Shanghai Science and Technology Innovation Action Plan Program (18411953800) and Shanghai Municipal Health Commission (2019).
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval Ethics Committee for Human Subject Research of the Shanghai Ji Ai Genetics and IVF Institute, Obstetrics and Gynecology Hospital, Fudan University (No: JIAI2017-06).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data may be obtained from a third party and are not publicly available.