Article Text

Abstract
Background The hallmark of the neurobehavioural phenotype of Williams-Beuren syndrome (WBS) is increased sociability and relatively preserved language skills, often described as opposite to autism spectrum disorders (ASD). However, the prevalence of ASD in WBS is 6–10 times higher than in the general population. We have investigated the genetic factors that could contribute to the ASD phenotype in individuals with WBS.
Methods We studied four males and four females with WBS and a confirmed diagnosis of ASD by the Autism Diagnostic Interview-Revised. We performed a detailed molecular characterisation of the deletion and searched for genomic variants using exome sequencing.
Results A de novo deletion of 1.55 Mb (6 cases) or 1.83 Mb (2 cases) at 7q11.23 was detected, being in 7/8 patients of paternal origin. No common breakpoint, deletion mechanism or size was found. Two cases were hemizygous for the rare T allele at rs12539160 in MLXIPL, previously associated with ASD. Inherited rare variants in ASD-related or functionally constrained genes and a de novo nonsense mutation in the UBR5 gene were identified in six cases, with higher burden in females compared with males (p=0.016).
Conclusions The increased susceptibility to ASD in patients with WBS might be due to additive effects of the common WBS deletion, inherited and de novo rare sequence variants in ASD-related genes elsewhere in the genome, with higher burden of deleterious mutations required for females, and possible hypomorphic variants in the hemizygous allele or cis-acting mechanisms on imprinting.
- autism spectrum disorders
- Williams-Beuren syndrome
- comorbidity
- exome sequencing
- neurobehavioural phenotype
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- autism spectrum disorders
- Williams-Beuren syndrome
- comorbidity
- exome sequencing
- neurobehavioural phenotype
Introduction
Williams-Beuren syndrome (WBS, OMIM#194050) is a rare neurodevelopmental disorder resulting from an heterozygous deletion of 25–27 genes at chromosome 7q11.23, estimated to affect approximately 1 in 7500 individuals.1 The WBS locus has a complex genomic architecture with a single copy region flanked by three large segmental duplications, each composed of three major blocks (A, B and C), located in the centromeric (c), medial (m) and telomeric (t) sides.2 Two types of recurrent rearrangements promoted by non-allelic homologous recombination (NAHR) can result in WBS syndrome. The most frequent is a 1.55 Mb deletion occurring between the medial and centromeric B blocks in 87% of cases. Around 10% of patients present a larger 1.83 Mb deletion due to a crossing over between centromeric and medial A blocks. The remaining 3% of patients with WBS show atypical deletions mediated by other mechanisms.3 4 The WBS multisystemic phenotype is characterised by cardiovascular disease, distinctive facies, connective tissue abnormalities and growth and endocrine alterations, among others.2 5 The main neurobehavioural hallmarks are mild to moderate intellectual disability (ID), hypersociability and relative language preservation.6 Interestingly, the reciprocal duplication of 7q11.23 (OMIM#609757) results in a phenotype with speech delay, language impairment, milder learning problems and clear social interaction deficits often associated with autism spectrum disorder (ASD).6 7
Due to their associated behavioural manifestations, ASD and WBS have often been described as diametric opposite disorders, although this consideration is an oversimplification of both phenotypes.8 9 Moreover, several cases of WBS with comorbid ASD have been reported and some authors have suggested that ASD features should be considered as part of the WBS phenotype.7–18 A recent meta-analysis of the comorbidity of ASD features in several well-defined genetic syndromes concluded that the prevalence of ASD features among WBS individuals is as high as 12%.19 Therefore, the frequency of ASD is 6–10 times higher in individuals with WBS than in the general population, a striking finding considering the typical WBS neurocognitive profile.
Potential modifiers of the common neurobehavioural phenotype of WBS include: (1) cis-acting mechanisms due to variable breakpoints altering flanking genes, although most patients described present common deletions of identical size3 17; (2) trans-acting factors present in the non-deleted hemizygous allele; (3) genetic mutations and/or structural variants elsewhere in the genome; (4) environmental events with or without epigenetic effects, including medical complications during development and early life. All these factors can have additive effects acting on a sensitised background caused by haploinsufficiency at the WBS locus.10 17 Two individuals with WBS and co-occurring ASD previously reported presented hyperserotonaemia and were homozygous for the short (s) allele in the promoter of the serotonin transporter SLC6A4 (5-HTTLPR), suggesting a possible modifier role of this locus.14 17
In the present work, we have investigated the genetic factors that could contribute to the ASD phenotype of eight individuals with WBS and comorbid ASD. We completed a detailed molecular characterisation of the deletion, genotyped the reported polymorphism at SLC6A4 and performed a genome-wide unbiased search of second-hits by exome sequencing following the strategy depicted in figure 1.

Strategy followed for the identification of second-hit genetic factors. DGV, Database of Genomic Variants; LoF, loss of function; MAF, minor allele frequency; SNV, single nucleotide variant.
Methods
Patient selection
From a cohort of 122 individuals with a diagnosis of WBS and confirmed 7q11.23 deletion by molecular techniques, we selected four males and four females aged 6–31 years with an associated diagnosis of ASD. The diagnosis was based on the direct observation by a trained psychologist and clinicians and confirmed in all of them using the Autism Diagnostic Interview-Revised (ADI-R). Written informed consent was obtained from all parents or legal caregivers.
7q11.23 deletion characterisation
Blood samples from probands and parents were obtained and genomic DNA was extracted using the Puregene DNA Purification Kit (Gentra Systems, Big Lake, Minnesota, USA). The size and parental origin of the deletion was established by the analysis of multiple ligation-dependent probe amplification and several single and multiple-copy microsatellites. Refined mapping of deletion breakpoints was also performed by quantitative analysis of paralogous sequence variants as previously described.3
SLC6A4 genotyping
The polymorphism in the serotonin transporter promoter SLC6A4 (5-HTTLPR) was genotyped by PCR and agarose gel electrophoresis, using primers previously described.17
Exome sequencing and analysis
Exomes were captured using the SureSelect Human All Exon V5 capture kit (Agilent, Santa Clara, California, USA) and libraries were sequenced on an Illumina MiSeq platform. Paired-end sequences were obtained with a read length of 250 bp.
Mapping, variant calling and filtering were performed using BWA and GATK’s standard parameters. The hg19 human genome reference version was used. Variant annotation was performed using ANNOVAR (http://www.openbioinformatics.org/annovar/), considering the variant frequency in control databases: dbSNP137 (http://www.ncbi.nlm.nih.gov/SNP/), ExAC (http://exac.broadinstitute.org/), Kaviar (http://db.systemsbiology.net/kaviar/) and an in-house database of 248 Spanish controls. The nature of the changes was assessed by PolyPhen and Condel (http://bg.upf.edu/fannsdb/) protein effect prediction algorithms.
For CNV detection, we applied ExomeDepth and compared our samples with a matched aggregate reference set of 248 in-house exomes captured and sequenced using the same protocol. CNVs were filtered based on their overlap with variants previously described in the Database of Genomic Variants and DECIPHER.20
Rare variant analysis and validation
We selected exonic variants with a minor allele frequency (MAF) lower than 0.002 according to several databases (previously mentioned) for heterozygous variants following a dominant inheritance model. For homozygous or compound heterozygous variants, under a recessive inheritance model, we selected a MAF ≤0.01. Since second-hit variants would act in the presence of a major hit and would not be expected to solely cause ASD, they might be present in the general population and inherited from unaffected progenitors. Moreover, given the high degree of genetic heterogeneity of ASD, they could affect hundreds of genes. Taking into account the prevalence of ASD in individuals with WBS and the large number of genes involved, we reasoned that the frequency of each individual variant should be relatively rare and, consequently, set our MAF threshold for homozygous or compound heterozygous variants at ≤0.01.
To validate variants and perform segregation studies in parental samples, we used Sanger sequencing by capillary electrophoresis (ABI PRISM 7900HT, Applied Biosystems, Foster City, California, USA). Primers were designed with the PRIMER3 program (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) and PCR reactions were carried following standard conditions.
Epistatic effects of rare variants
To study the putative contribution of epistatic effects of rare SNVs on gene expression deregulation in WBS, we compiled a list of genes previously altered in WBS by various mechanisms, including transcriptional dysregulation, differential methylation and the direct GTF2I targets. We performed a systematic literature review and selected only high-quality studies done in human subjects, obtaining 2251 candidate genes (online supplementary table 1).21–24
Supplemental material
Analysis of ASD susceptibility loci
To study the potential contribution of common variants, we selected loci previously associated with ASD by Genome-Wide Association Studies (GWAS). Variants containing the term ‘Asperger’ or ‘Autism’ were extracted from the Genome-Wide Repository of Associations between SNPs and phenotypes (GRASP) database V.2.0 (online supplementary table 2).25 Allele frequency in the WBS cohort was then compared with non-Finnish European (NFE) population data from ExAC by Fisher’s exact test and q-value and false discovery rate (FDR) values were calculated using the R package qvalue.26 To avoid population stratification, ExAC allele frequencies were compared with that reported in Spanish Variant Server, including data from 578 Spanish individuals (http://csvs.babelomics.org/).
Results
Clinical characteristic of selected patients
The study was performed in eight Spanish patients with WBS (four males and four females), which had periodic follow-up in a multidisciplinary clinic and complete neurobehavioural evaluation due to a comorbid diagnosis of ASD. Their main clinical characteristics are summarised in table 1. All patients fulfilled criteria in the social interaction and restrictive behaviour domains while two of them (WBS2 and WBS3) did not reach the verbal communication domain threshold. Family history of ASD or psychiatric disorders was positive on the paternal side in two cases (WBS3 and WBS6) and negative in the others (table 1).
Clinical characteristics of the WBS individuals with associated ASD
Detailed characterisation of the 7q11.23 deletion
To define potential cis-acting factors in the deleted allele, we characterised all deletions at the molecular level including determination of the parental origin. All rearrangements were de novo and all but one (7/8) were of paternal origin (table 2). The most common 1.55 Mb deletion was found in 6/8 patients, whereas 2/8 carried the larger recurrent 1.83 Mb deletion mediated by NAHR between A blocks (figure 2). In two patients (WBS1 and WBS3), the common 1.55 Mb deletion had been mediated by an inversion in the transmitting progenitor.
Summary of the main genetic findings per patient

Schematic representation of the Williams-Beuren syndrome (WBS) locus, showing the two most common deletions and the gene content of the B block. The B block contains three genes: GTF2I, NCF1 and GTF2IRD2. Whereas GTF2I and NCF1 have a single functional copy located at the medial B block, GTF2IRD2 has two functional copies, located at the medial (GTF2IRD2) and telomeric (GTF2IRD2B) B blocks. The 1.55 Mb deletion is mediated by B blocks and results in a chimeric medial-centromeric block, with the number of functional copies of GTF2IRD2 and NCF1 depending on the deletion breakpoint. In contrast, the 1.83 Mb deletion is mediated by A blocks, resulting in the loss of the medial and centromeric B blocks with functional copies of GTF2IRD2 and NCF1.
Since GTF2IRD2 has been postulated as a possible modulator of WBS cognitive phenotype,27 we analysed the deletion breakpoints to assess the number of functional copies of this gene in the patients (figure 2). As expected, the deletions mediated by A blocks included the entire medial B block with a copy of GTF2IRD2, resulting in a loss of one functional copy (1M/2T: 1 medial (GTF2IRD2)/2 telomeric (GTF2IRD2B)). The two patients with inversion-mediated deletions had a 1M/3T genotype, originated from the loss of a medial copy and a gain of a telomeric copy of GTF2IRD2. Among the patients with 1.55 Mb deletions, three had breakpoints before GTF2IRD2, with no change in the number of functional copies (2M/2T), whereas in one patient the breakpoint occurred within GTF2IRD2, creating a chimeric copy (Ch) between the medial gene and the centromeric pseudogene (1Ch/1M/2T) (table 2). The breakpoints also affect the number of NCF1 copies, by either deleting or not one of the functional copies.
Hemizygous variants in the 7q11.23 region
To look for trans-acting factors in the 7q11.23 allele present in hemizygosity, we analysed the entire captured region within the WBS common deletion locus (chr7:72 700 000–74 250 000) looking for over-represented rare and common variants, shared haplotypes and rare deleterious SNVs. For over-represented variants, we compared allele frequencies of all described hemizygous variants (n=32) in our cohort to those reported in ExAC for European population. Variants with significantly different frequencies in ExAC and Spanish Variant Server were excluded to avoid population stratification. Due to the small sample, none of the variants reached statistical significance, but we identified a nearly significant association (p=0.076) with rs12539160, which was present in two individuals (WBS2 and WBS6). This synonymous variant located near an exon-intron boundary of MLXIPL had been previously associated with ASD in a GWAS.28 Taking advantage of the hemizygosity of SNPs in the deleted single-copy region, we extracted phased haplotypes in this interval to study if a common haplotype was shared between individuals. Two linkage disequilibrium blocks were identified from rs1128349 to rs13227841 (DNAJC30 to WBSCR28) and from rs17851629 to rs2074667 (located in GTF2IRD1). Allele frequencies of the tag markers did not differ significantly from those in the general population and no common shared haplotype was identified in our cohort. In addition, we looked for rare SNVs in the single-copy region of the WBS locus. Only hemizygous variants with a MAF <0.01 were selected. After filtering for exonic variants and excluding synonymous SNVs with no functional effect, we remained with two non-synonymous variants in genes MLXIPL and TBL2 predicted as tolerant by various protein effect prediction algorithms ((SIFT (sorting intolerant from Tolerant), PolyPhen (Polymorphism Phenotyping) and Condel)). No rare variants were identified at GTF2I, NCF1 and GTF2IRD2, the genes in the flanking segmental duplications.
Genome-wide analysis of rare variants
Copy number variants
We also studied the presence of additional rearrangements that could explain the autistic symptoms in our cohort. In addition to the WBS 7q11.23 deletion, we observed an average of 25 CNVs per patient ranging from 170 bp to 334 kb. None of the additional rearrangements overlapped with known genomic disorders or was previously associated with neurodevelopmental disorders and all overlapped with previous CNVs described in the general population.29 Only two CNVs comprised ASD candidate genes (SIK1 and DUSP22) (online supplementary table 3).30 The CNV involving SIK1 was a partial duplication affecting the last two exons of the gene, paternally inherited in patient WBS3. The heterozygous deletion completely containing DUSP22 was found in patient WBS7 and not in her mother, but paternal sample was unavailable.
Rare single nucleotide variants
We prioritised deleterious variants present in a list of candidate genes from -Simons Foundation Autism Research Initiative (SFARI) (n=791) (online supplementary table 3) and/or highly constrained genes, as previous studies have shown that genes predisposing to ASD carry a low burden of disrupting mutations in the general population.30 31 Constrained genes were defined as those with a probability of being loss of function (LoF) intolerant (pLI >0.9) according to ExAC.32 LoF and missense variants predicted as damaging by both SIFT and PolyPhen were considered deleterious variants.
We detected a total of 38 rare deleterious SNVs in 34 candidate genes (online supplementary table 4). All LoF variants (n=6) were validated and segregation studies were performed on all samples, showing a de novo variant in the UBR5 gene in case WBS5 (table 2). De novo variants in the UBR5 gene, two missense and one LoF, have been previously described in ASD individuals.33–35 WBS5 is a severely affected female who presented an obsessive ritualistic behaviour, hypersensitivity to noise and physical stimuli, had not developed language and did not show eye contact.
As for highly constrained genes, excluding candidate genes, we detected 68 SNVs in 65 genes, including four LoF mutations (table 2). All genes selected on the basis of being LoF intolerant but harbouring a LoF in our cohort were brain-expressed and some had been previously associated with psychiatric and neurodevelopmental disorders, such as SEC24C, CXXC1 and EPHB1, which was mutated in two patients with WBS.36–39
Epistatic effects of rare variants
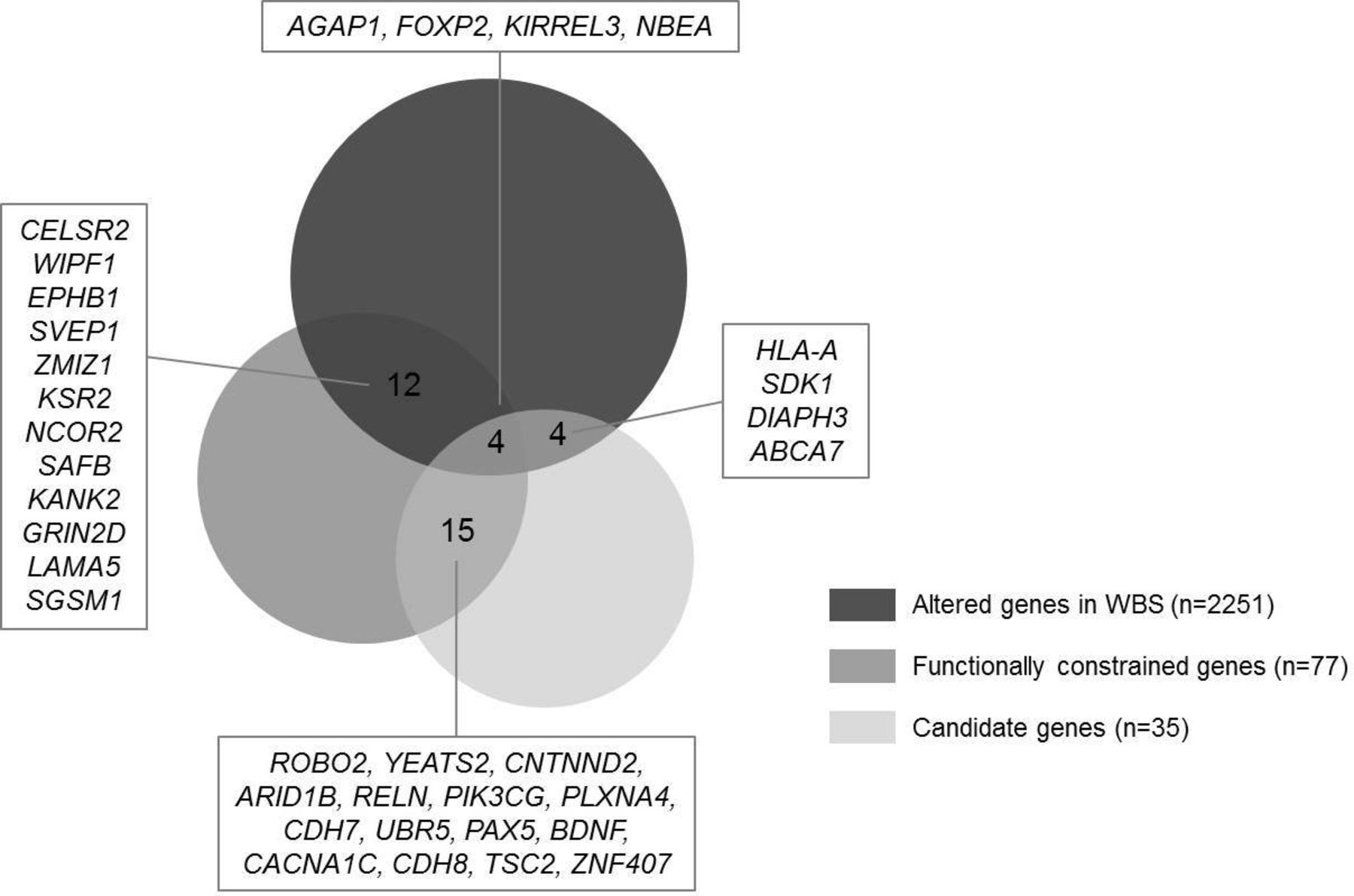
To study if second-hit variants could be disrupting genes already altered in WBS and act by an epistatic effect, we intersected a manually curated list of genes with altered expression in WBS with the list of mutated genes (figure 3). The results showed that 20 of mutated genes were also altered in WBS. Two of them harboured a LoF in our cohort: AGAP1 and EPHB1 in two cases.

Overlap between candidate and functionally constrained genes mutated in our cohort and genes altered in Williams-Beuren syndrome (WBS).
Burden of rare deleterious mutations, increased in females
In total, six patients with WBS with ASD carried, in addition to the specific de novo 7q11.23 deletion, one to three strong candidate rare genetic variants (CNV and/or SNV) that could contribute to the ASD phenotype, either inherited or de novo (table 2).
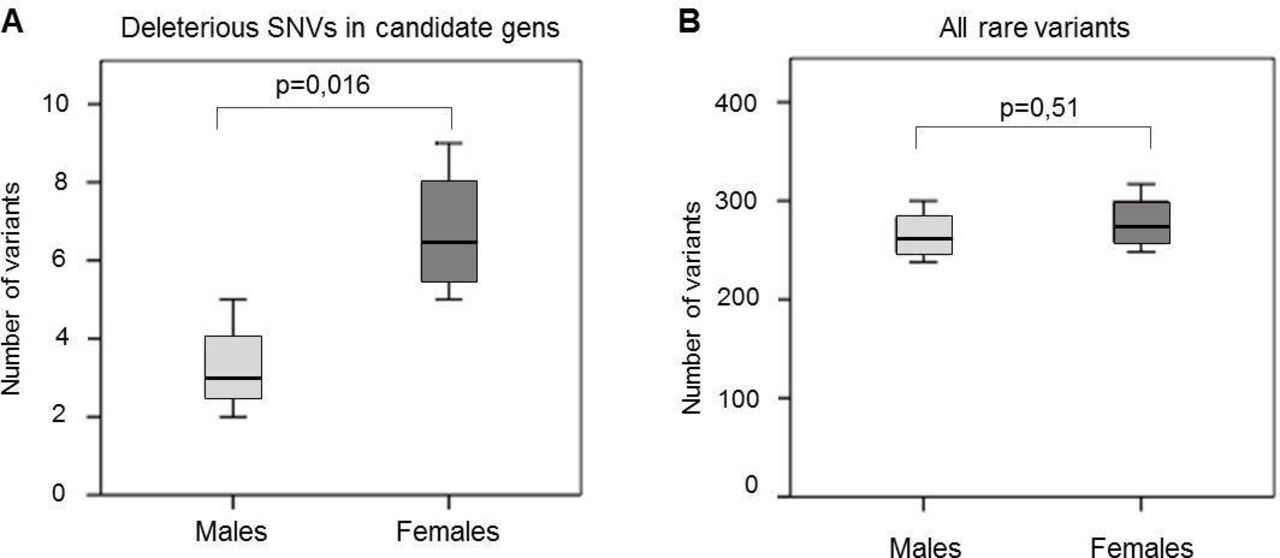
Since ASD is four times more prevalent in males than females, it has been suggested that females have a higher risk threshold and require a higher genetic load to develop the disorder.40 To examine if females in our cohort carried a higher genetic burden, we compared the frequency of rare deleterious variants in ASD candidate genes between males (n=4) and females (n=4) (figure 4). The results showed a statistically significant increase (p=0.016) in the number of total mutations per patient in females ( , SD=1.7) compared with males (
, SD=1.7) compared with males ( , SD=1.2). This effect was not seen when comparing the burden of all rare variants between genders (p=0.51).
, SD=1.2). This effect was not seen when comparing the burden of all rare variants between genders (p=0.51).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Gender (male vs female) comparison of the average number of rare variants in Williams-Beuren syndrome-autism spectrum disorders cases. Statistical significance in all tests was calculated using a two-sided Student’s t-test, excluding SNVs of the X and Y chromosomes from the analysis. (A) Deleterious variants in candidate genes. (B) All rare variants. SNV, single nucleotide variant.
Association study with candidate ASD genes
To study the potential role of common variants, we analysed the loci previously associated with ASD and compared their variation frequency in our cohort with that of NFE ExAC population. A total of 645 single-nucleotide markers located in coding regions were analysed, of which only 13 obtained a significant p value (p<0.05) and none passed FDR correction. For 10 of the significantly over-represented SNVs in our cohort, the over-represented allele corresponded to the risk allele, whereas it was the protector allele for the 3 remaining. Three of the markers had been previously described as Expression Quantitative Trait Loci (eQTL) in brain, with rs2275477 associated with increased expression of OSCP1 and rs4823086 and rs5749088 with transcript RP1.130H16.16/CCDC157.41 We also identified a nominally significant increase in the frequency of rs2135720, a non-synonymous SNP within PCDH15 associated with lipid traits.42
Finally, we also genotyped two promoter variants at SLC6A4 not captured in exome data that had been proposed as modifiers for the phenotypic outcome in WBS in two patients with autistic symptoms and hyperserotonaemia. In our cohort, the genotype frequencies were similar to those found in population of European origin, with 3/8 individuals homozygous for the major long variant (l), 4/8 presenting an heterozygous genotype (l/s) and 1/8 individual being homozygous for the s allele.
Discussion
We have performed a genome-wide comprehensive analysis to investigate possible genetic factors in eight individuals presenting with both diagnoses of WBS and ASD. To date, the analysis of exome data failed to reveal genetic variants that could explain the variance of the social behaviour phenotype in a cohort of 85 patients with WBS without comorbid ASD,43 and the only locus suggested to act as a modifying factor was the serotonin transporter SLC6A4 (5-HTTLPR), based on two individuals with WBS and ASD who had hyperserotonaemia and were homozygous for the ss polymorphism at the gene promoter.17 However, the results in our cohort show genotype frequencies similar to those of the general population and do not support a role of SLC6A4.
The characterisation of the deletion showed no atypical rearrangements and different breakpoints in the patients, excluding the alteration of flanking genes as a major cause. In the studied patients the deletion had originated in the paternal allele in seven of eight, suggesting that epigenetic control mechanisms could influence ASD risk in WBS. This parental bias was not present in our larger cohort of WBS without comorbid ASD (n=374 trios) as the origin of the deletion was 50% maternal and 50% paternal. There are some evidences supporting the imprinting of the 7q11.23 region based on expression of FKBP6 and GTF2I.44–46 However, in a recent study of six patients with WBS and ASD, the deletion was of maternal origin in four out of six patients.47 Collectively, there is suggestive evidence regarding a possible role of genetic imprinting, but not enough data to confirm it, which demands characterisation of the deletion and parental origin in additional individuals with WBS and ASD to confirm whether imprinting can influence phenotypic variability.
Besides deletion origin and breakpoints, hemizygous variants unmasked in the deleted region could also act as second-hit modifying factors. The detailed analysis of SNVs did not reveal any common haplotype or any rare variant in the coding regions of GTF2I or GTF2IRD2. However, the analysis of common variants revealed an over-representation of a relatively rare hemizygous variant previously associated with ASD (rs12539160).28 It has a MAF of 0.06 in European population and of 0.03 in the Spanish population but it was found in two patients (0.25) of our cohort, representing an eightfold increase in frequency.32 While not meeting strict criteria for statistical significance (p=0.076), this finding deserves additional investigation in future studies. In a recent study, exome data of 85 patients with WBS was analysed in order to search for common and rare variants that could account for the variance in the social behaviour of these patients.43 Together with the genotyping of our cohort of non-autistic WBS, this variant (rs12539160) was found with a frequency of 0.0573 among 157 WBS individuals which is similar to general population. Although this variant lies in the intronic region of MLXIPL, mainly involved in lipid abnormalities, deregulation of the same or other nearby gene either by this SNP or other risk variants in linkage disequilibrium not detected by exome sequencing might contribute to ASD risk in WBS.
We also studied genetic variation at a genome-wide level. Regarding CNVs, no additional large (>500 kb) or pathogenic rearrangement was identified. Likewise, previous studies evaluating second-hit CNVs in genomic disorders showed that additional rearrangements were more frequent in disorders of variable expressivity than in syndromic entities. In fact, only 5% of individuals with WBS carried a second event, similar frequency than in the control population, suggesting that additional large CNVs may cause a more severe phenotype and/or be incompatible with life.48 However, relatively small CNVs altering ASD candidate genes and therefore potential contributors to the ASD phenotype were found in two patients, a partial duplication of the last two exons of SIK1, and a complete heterozygous deletion of DUSP22.
The analysis of rare deleterious SNVs on ASD candidate and functionally constrained genes uncovered several brain-expressed genes that harbour LoF variants in our cohort. Among those, we detected a de novo stop mutation affecting UBR5, an E3 ubiquitin-protein ligase in a severely affected female. Therefore, the presence of WBS and ASD in this individual is probably due to the co-occurrence of two mechanistically unrelated de novo events, the WBS deletion and the UBR5 mutation.
However, the increased prevalence of ASD features in WBS and the low frequency of de novo events suggest that other genetic factors of smaller effect may be influencing the ASD risk in most cases. In fact, we have identified several rare variants in ASD candidate genes. Two of the genes harbouring LoF mutations, EPHB1 and AGAP1, have been found deregulated in WBS, suggesting a possible epistatic effect. AGAP1 was hypermethylated in patients with WBS compared with individuals with the reciprocal duplication, which is associated with a phenotype of language impairment, anxiety and increased risk of ASD and schizophrenia.24 49–51 EPHB1 was underexpressed in patients with WBS and individuals with atypical deletions and low IQ. Inherited mutations in those genes would result in further deregulation of the expression already altered by the WBS deletion21 and higher risk for a more severe phenotype.
Globally, six of the eight patients with WBS with ASD carried, in addition to the specific de novo deletion at the 7q11.23 locus, one to three rare genetic variants (CNV and/or SNV) altering candidate genes that could contribute to the ASD phenotype. Consequently, a proportion of cases of WBS with associated ASD could be explained by the same factors influencing ASD risk in the general population with a threshold model. Given the increased risk for ASD in WBS, the WBS deletion would act as a predisposing factor facilitating the role of other genetic (inherited or de novo) and/or environmental factors by acting on an already sensitised background.
Interestingly, the average number of rare deleterious variants in candidate genes was significantly higher in females than males, an effect not observed when considering all rare variants. Although our small sample size requires caution interpreting the results, this difference could support the higher risk threshold requiring higher genetic load in females to develop ASD, or could just be explained by the fact that females in our cohort had a more severe presentation than males. Further studies assessing the difference in prevalence and severity of ASD features between genders in individuals with WBS will help clarify if the female protective effect has a role in the expressivity of disorders of full penetrance.
Finally, we assessed the contribution of common variation by looking for over-represented variants previously associated with ASD. Although none of the variants passed FDR correction, three markers with nominal p values had been previously described as eQTLs in brain and may have a direct functional effect. However, our study is limited by the fact that most common variation is not covered by exome sequencing, as it resides in non-coding regions. Currently, common variation seems to explain at least 20% of ASD liability.52 Therefore, further studies regarding the role of common variants in autism will provide a necessary basis for future studies.
In summary, our work represents a thorough assessment of second-hit modifier genetic factors in individuals with WBS and associated ASD. Similar to previous reports, patients did not differ in deletion breakpoints, discarding the role of atypical rearrangements. However, in seven of eight the deletion had originated in the paternal allele, a factor that had not been addressed in previous studies. In addition, two individuals carried a relatively rare hemizygous variant previously associated with ASD, representing an eightfold increase with respect to the general population. Inherited rare variants and a de novo nonsense mutation in ASD-related or functionally constrained genes were identified in six cases, with higher burden in females compared with males. Taken together, these results suggest that imprinting mechanisms, trans-acting factors in the remaining allele and inherited or de novo rare sequence variants elsewhere in the genome may play a role in the susceptibility to ASD in WBS. Therefore, similar factors influencing ASD risk in the general population also shape phenotypic variability in disorders with full penetrance.
Acknowledgments
The authors would like to thank the patients and their families for their support. The authors would also like to thank Marcos López-Sánchez for his help with the data acquisition and handling.
References
Footnotes
MC-S and MC-R are joint first authors.
Contributors MC-S and MC-R analysed the exome data, performed the experiments and drafted the manuscript. DP-G and MGP-V contributed with the clinical and phenotypic information. RF contributed with detailed molecular characterisation of deletions. IC and LAP-J conceived the study and participated in the design and data interpretation, and helped in drafting the manuscript. All authors read and approved the manuscript.
Funding This work was funded by grants from the Spanish Ministry of Economy and Competiveness (FIS PI16/00369 and PI1302481 cofunded by FEDER, and ‘Programa de Excelencia María de Maeztu’ MDM-2014-0370), and the Generalitat de Catalunya (2017SRG01974 and ICREA-Acadèmia program). MC-R had a predoctoral fellowship of Ministry of Education, Culture and Sport (FPU16/06907) and DP-G had a predoctoral fellowship from the Instituto de Salud Carlos III (FI11/00656).
Competing interests LAP-J is scientific advisor of qGenomics SL. The remaining authors declare no conflict of interest.
Patient consent for publication Not required.
Ethics approval The study was approved by the Clinical Research Ethics Committee of the Parc Salut Mar.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.