Article Text

Abstract
Background Gene panel testing has become the norm for assessing breast cancer (BC) susceptibility, but actual cancer risks conferred by genes included in panels are not established. Contrarily, deciphering the missing hereditability on BC, through identification of novel candidates, remains a challenge. We aimed to investigate the mutation prevalence and spectra in a highly selected cohort of Greek patients with BC, questioning an extensive number of genes, implicated in cancer predisposition and DNA repair, while calculating gene-specific BC risks that can ultimately lead to important associations.
Methods To further discern BC susceptibility, a comprehensive 94-cancer gene panel was implemented in a cohort of 1382 Greek patients with BC, highly selected for strong family history and/or very young age (<35 years) at diagnosis, followed by BC risk calculation, based on a case–control analysis.
Results Herein, 31.5% of patients tested carried pathogenic variants (PVs) in 28 known, suspected or candidate BC predisposition genes. In total, 24.8% of the patients carried BRCA1/2 loss-of-function variants. An additional 6.7% carried PVs in additional genes, the vast majority of which can be offered meaningful clinical changes. Significant association to BC predisposition was observed for ATM, PALB2, TP53, RAD51C and CHEK2 PVs. Primarily, compared with controls, RAD51C PVs and CHEK2 damaging missense variants were associated with high (ORs 6.19 (Exome Aggregation Consortium (ExAC)) and 12.6 (Fabulous Ladies Over Seventy (FLOSSIES)), p<0.01) and moderate BC risk (ORs 3.79 (ExAC) and 5.9 (FLOSSIES), p<0.01), respectively.
Conclusion Studying a large and unique cohort of highly selected patients with BC, deriving from a population with founder effects, provides important insight on distinct associations, pivotal for patient management.
- hereditary breast cancer
- BRCA1
- BRCA2
- PALB2
- NGS
- gene panel
- genetic testing
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Loss-of-function (LoF) variants in the highly penetrant genes, BRCA1 and BRCA2, are the major players for breast and ovarian cancer susceptibility; individuals carrying such pathogenic variants (PVs) face an increased lifetime risk for cancer diagnoses.1 The implementation of BRCA1 and BRCA2 genetic testing into clinical practice has enabled the identification of individuals at high risk and the application of tailored management guidelines, significantly improving both cancer prevention and survival.2
Mutations in BRCA1/2 genes account for 5%–10% of breast cancer (BC) cases. This can be slightly higher in populations with strong founder effects, such as the Ashkenazi Jewish, or to a lesser extent, Greek.3–6 Testing for BRCA1/2, for more than two decades, revealed an important limitation; a substantial number of individuals with early diagnosis and/or strong family history receive a negative test result. Further investigation on high-risk families, along with advances in sequencing technologies, have led to the characterisation of additional BC predisposition alleles in genes, most of which encode for proteins involved in DNA repair through homologous recombination.7
Nowadays, a number of high penetrant genes, that is, TP53 and PALB2, genes of intermediate penetrance, that is, CHEK2 and ATM, as well as syndromic genes, that is, CDH1, PTEN and STK11, are included in gene panels. Alongside, multigene panel testing has replaced, to a large extent, traditional single gene testing, where additional genes, PVs of which predispose for other types of cancers or are low penetrant, are analysed. In this context, the probability of receiving an unclear or inconclusive result increases significantly, while the referring clinician’s major concern remains the clinical actionability of the genetic testing results, mainly due to lack of defined management guidelines.8
We therefore sought to investigate the mutation prevalence and spectra in a highly selected cohort of Greek patients with BC diagnosed at a very young age (<35 years) and/or with strong family history, implementing a comprehensive 94 gene panel, while calculating gene-specific BC risks. Both the extensive number of genes questioned, most of which are implicated in cancer predisposition and DNA repair and the stringent patient selection criteria set, provide important benefits for identification of novel BC candidates and revelation of important associations to BC susceptibility.
Patients and methods
Patient selection
The study cohort included 1382 Greek patients with BC having strong family history (at least three cancer diagnoses of breast, ovarian or pancreatic cancer from the same side of the family) and/or diagnosed <35 years. All patients were index, were not related and have been retrospectively selected from referrals to Molecular Diagnostics Laboratory of National Centre for Scientific Research (NCSR) ‘Demokritos’ between the years 1999 and 2017. Written informed consent was obtained from all individuals prior to genetic testing. The study was in agreement with the 1975 Helsinki statement.
Testing for Greek BRCA1 founder alleles
Initial screening involved testing for the five Greek founder and one recurrent BRCA1 mutations, as previously described.3 9 This includes (1) three BRCA1 large genomic rearrangements (LGRs), all of which disrupt the BRCA1 C-terminus domain and are confined to the Greek population4 5 and (2) three single nucleotide variants, namely, c.5212G>A, c.5266dupC and c.5251C>T0.3
Genomic capture and massively parallel sequencing using Trusight cancer panel
Germline DNA is enzymatically fragmented, adaptor tagged, indexed and captured to target the 1736 genomic regions of 94 cancer predisposing genes using TruSight Cancer Panel, following manufacturer’s instructions (Illumina, San Diego, USA). The complete gene list is shown in the online supplementary table 1.
Supplemental material
Amplified libraries were evaluated qualitatively and quantitatively using Fragment Analyzer (Advanced Analytical Technologies, Heidelberg, Germany). Indexed libraries were sequenced on MiSeq using the Standard V2 kit performing 150 base paired-end reads, while FASTQ, BAM and VCF files were generated through Illumina MiSeq Reporter; annotation was performed against the human reference genome GRCh38 using VariantStudio V.3 (Illumina). The minimum base and amplicon coverage were 50×, and 100×, respectively, while the mean read depth was 182×. All PVs were confirmed by Sanger sequencing.
Multiplex ligation-dependent probe amplification (MLPA)
Detection of large deletions and duplications among the genes tested, excluding the Greek founder BRCA1 LGRs, was not sufficiently reliable through NGS-based testing. Therefore, SALSA MLPA kit P002, P045, P190 and P056 were used to assess LGRs involving BRCA1, BRCA2, CHEK2 and TP53 genes, respectively, following manufacturer’s instructions (MRC-Holland, Amsterdam, the Netherlands).
Evaluation of missense variants
Missense variants identified herein have been evaluated and classified based on data deriving from: (1) their characterisation based on in vivo and/or in vitro functional assay(s), (2) classification based on ClinVar10 and (3) prediction models (SIFT, PolyPhen, AlignGVGD, PastCons, Phylop and Mutation Taster), providing (1) strong and (2, 3) supporting evidence, respectively, based on the American College of Medical Genetics and Genomics (ACMG) classification guidelines.11 Detailed description of the rare missense variants classified as pathogenic is illustrated in the online supplementary table 2.
Supplemental material
Statistical analysis and case–control study
Quantitative variables are presented as mean±1 SD, while categorical variables as frequency. Non-parametric Wilcoxon rank-sum test was used to compare age association between mutations per gene. Comparisons of categorical variables between groups were performed using Pearson’s χ2 test. ORs were calculated using the Fisher’s exact test. P values were corrected for multiple comparison using false discovery rate method and were considered statistically significant when <0.05.
Cumulative LoF variants per gene were directly compared with the non-The Cancer Genome Atlas (TCGA) exomes, non-Finnish European (NFE) Exome Aggregation Consortium (ExAC) data set,12 and, when applicable, to the Fabulous Ladies Over Seventy (FLOSSIES) European-American data set (https://whi.color.com/). ExAC–NFE variants with allele frequency >0.2% and low-quality variants were filtered out. For both data sets, only pathogenic/likely PVs by ClinVar were included. Individuals carrying LGRs, double LoF variants and the low-risk CHEK2 variants: p.(Ile157Thr) and p.(Ser428Phe) were excluded from analyses.
Two separate analyses were performed for TP53 mutations, one including all carriers and one for those with BC diagnosis <35 years.
Results
Prevalence of LoF variants
The detection rate of LoF variants among the 1382 patients with BC with strong family history and/or young age at BC diagnosis (<35 years) tested was 31.5% (436/1382). In total, 446 LoF variants have been identified in 436 individuals in 28 genes and more specifically, BRCA1, BRCA2, ATM, BLM, BRIP1, CDKN2A, CHEK2, DICER1, ERCC3, FANCC, FANCD2, FANCI, FANCL, FANCM, MLH1, MSH6, NBN, NF1, PALB2, PMS2, PTEN, RAD51C, RAD51D, RECQL4, SDHB, SDHC, SLX4 and TP53. BRCA1/2 LoF variants dominated the variant spectra, identified in 24.8% (343/1382) of the patients tested. BRCA1 Greek founder variants accounted for approximately half (11.4%; 158/1382) of the BRCA1/2 variants identified. A detailed list of the variants, along with patients’ age at BC diagnosis, personal and family history are illustrated in the online supplementary table 3.
Supplemental material
A considerably high number of the tested patients (6.7%; 93/1382) harbour PVs in known, suspected or candidate BC genes, other than BRCA1/2. Detected mutations scatter among 26 genes with frequencies recorded as follows: 22 in CHEK2 (1.6%), 18 in ATM (1.3%), 11 in PALB2 (0.8%), eight in each of TP53 and RAD51C (0.6%), four in RAD51D (0.3%), three in each of PTEN, MSH6 and NBN (0.2% each), two in each of BRIP1, ERCC3, FANCL, FANCM, PMS2 and SLX4 (0.15% each) and one in each of BLM, CDKN2A, FANCC, FANCD2, FANCI, RECQL4, DICER1, MLH1, NF1, SDHB and SDHC (0.07%). Interestingly, the detection rate of PVs in established cancer genes alone, that is, BRCA1, BRCA2, TP53, PTEN, PALB2, ATM and CHEK2 is 29.3%. Noteworthy, 16 missense variants in five genes, namely, BRCA1, BRCA2, CHEK2, TP53 and CDKN2A, were classified as pathogenic, based on the detailed evaluation for variant classification, following ACMG guidelines (online supplementary table 2). A graphical illustration of the distribution of damaging variants among the 28 genes identified in this study is shown in figure 1.

Distribution of loss-of-function variants, in known, candidate and suspected breast cancer predisposing genes.
Excluding BRCA1/2 and based on current knowledge, LoF variants in genes conferring high BC risk, that is, in PTEN, PALB2 and TP53, were identified in 1.6% of the patients tested. Damaging variants in genes associated with moderate (ATM and CHEK2) and suspected BC risk (SLX4, NBN, BLM, ERCC3, FANCM, RAD51C, RAD51D, MSH6, PMS2, MLH1 and NF1) were identified in 2.9% and 2.2% of the patients tested, respectively. Variants in candidate BC genes, proteins of which are either involved in the Fanconi anaemia pathway (FANCC, FANCD2, FANCI, FANCL and BRIP1) or in genes associated with known syndromes (SDHB, SDHC, CDKN2A, RECQL4 and DICER1), were detected in 0.85% of the tested individuals (figure 2).
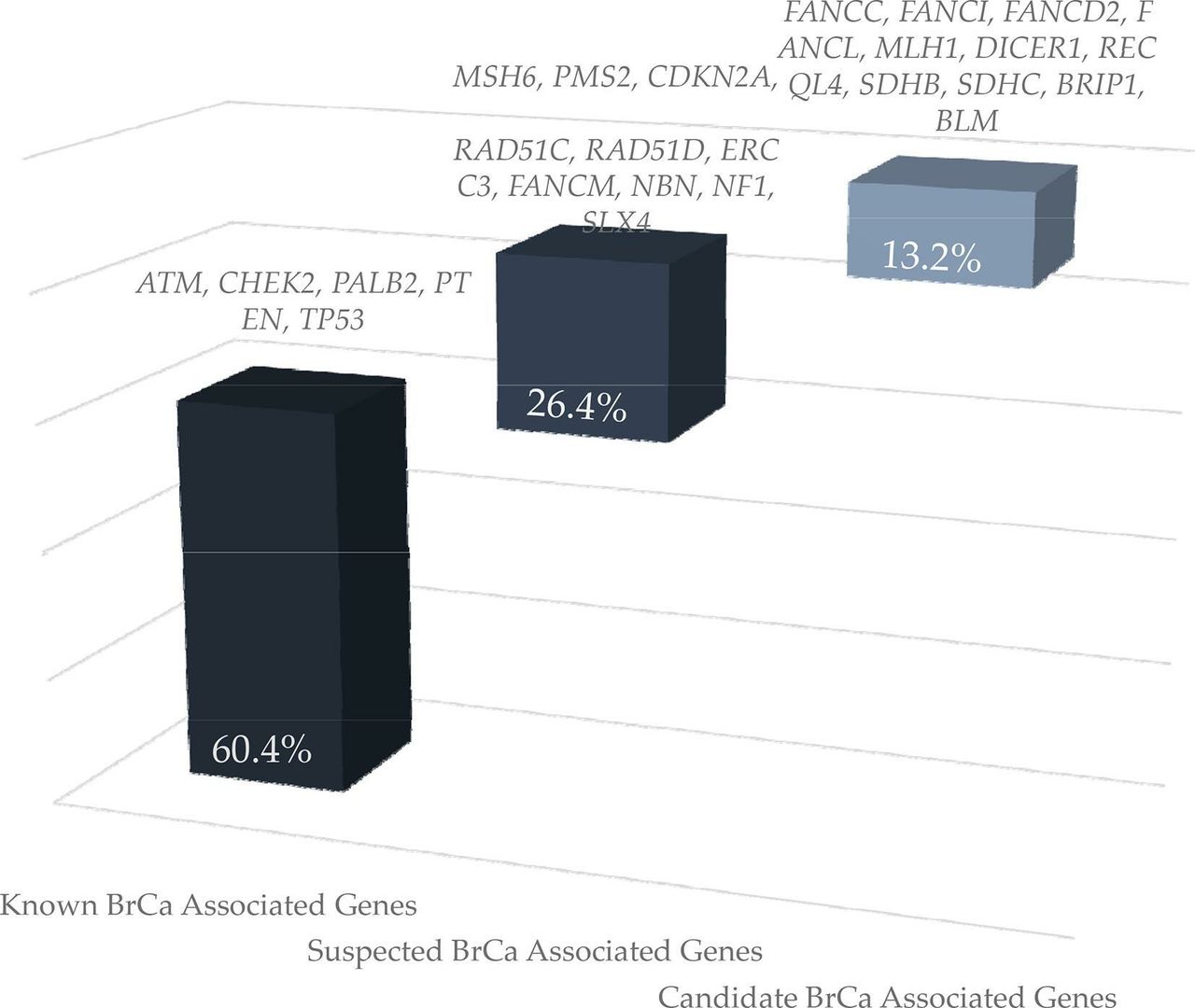
Loss-of-function variant distribution in known, suspected or candidate genes associated with breast cancer, other than BRCA1 and BRCA2.
LGRs were evaluated through non-NGS experimentation, in four genes namely, BRCA1, BRCA2, TP53 and CHEK2. In total, 5.7% (79/1382) of the patients in our cohort carried LGRs, with only 0.7% (10/1382) of them being different from the Greek BRCA1 founders. These were one BRCA1 deletion affecting the non-coding exon 1 and coding exon 2 (based on LRG_292), two BRCA2 deletions (affecting exons 12 and 13 and exon 3 (based on LRG_293)), three novel CHEK2 deletions (affecting promoter and exon 1, exon 6 and exons 2 and 3 of the gene (based on LRG_302)) and a TP53 deletion of the promoter and the non-coding, exon 1 of the gene (based on LRG_321).
In addition to the LoF variants, the low-risk BC variant, CHEK2 c.470T>C and the hypomorphic BRCA2 missense variant, c.9104A>C, were detected in 12 and two individuals, respectively, representing a frequency of 0.9% and 0.15%, respectively.
Case–control analysis for breast cancer risk estimations
Associations between detected LoF variants per gene were compared with controls extracted from the ExAC (non-Finnish/non-TCGA) and FLOSSIES (European-American) data sets.
TP53 deleterious variants were associated with high BC risk (OR 4.79; 95% CI 1.77 to 11.2, p<0.01 and OR 11.2; 95% CI 2.87 to 75.64, p<0.01) when compared with ExAC and FLOSSIES controls, respectively. PALB2 LoF variants were also significantly associated with high BC risk (OR 8.03; 95% CI 2.81 to 20.42, p<0.01 and OR 6.3; 95% CI 1.81 to 22.75, p<0.01) when compared with ExAC and FLOSSIES controls, respectively. Interestingly, pathogenic RAD51C variants showed a significant association to high BC risk (OR 6.19; 95% CI 2.23 to 15.03, p<0.01 and OR 12.6; 95% CI 2.87 to 75.64, p<0.01) when compared with ExAC and FLOSSIES controls, respectively, while ATM deleterious variants were significantly associated with moderate BC risk (OR 3.41; 95% CI 1.87 to 5.86, p<0.01 and OR 4.12; 95% CI 2.0 to 8.3, p<0.01) when compared with ExAC and FLOSSIES controls, respectively.
Intriguingly, the association of LoF CHEK2 variants neither was associated with clinically relevant BC risk nor was statistically significant when compared with ExAC (OR 1.66; 95% CI 0.98 to 2.67, p=0.11), while was associated with moderate BC risk, when compared with FLOSSIES (OR 2.67; 95% CI 1.44 to 4.68, p<0.01). When performing a subanalysis that included only missense LoF CHEK2 variants, not only reached statistical significance but also provided clinically relevant associations to moderate BC susceptibility (OR 3.79; 95% CI 1.86 to 7.12, p<0.01) when compared with ExAC and moderate to high BC susceptibility (OR 5.9; 95% CI 2.38 to 14.78, p<0.01) when compared with FLOSSIES.
The combined prevalence of LoF variants per gene (<7 variants), along with ORs for BC risk between selected Greek patients with BC patients and controls from ExAC and FLOSSIES are summarised in table 1, while a detailed table of ORs derived from the case–control study for all genes is listed in the online supplementary table 4. Therefore, ORs derived from the case–control analysis for only CHEK2, ATM, PALB2, TP53 and RAD51C are presented on a Forest plot diagram in figure 3.
Supplemental material
Prevalence and ORs for breast cancer risk of LoF variants in genes between selected Greek patients with breast cancer and reference controls from ExAC and FLOSSIES.

{kind=link}
{kind=link}
{kind=link}
Forest plot illustrating OR for breast cancer between selected Greek patients with breast cancer and controls from ExAC and FLOSSIES. Notes: CNV carriers, double LoF variant carriers and the low risk CHEK2 variants: p.(Ile157Thr) and p.(Ser428Phe) were excluded from the analysis. Genes that had at least seven LoF variants were included. ExAC data extracted were non-TCGA/non-Finnish Europeans.FLOSSIES data extracted were European-Americans. ExAC, Exome Aggregation Consortium; FLOSSIES, Fabulous Ladies Over Seventy; LoF, loss of function; miss, missense; TCGA, The Cancer Genome Atlas.
Families with damaging variants in established hereditary cancer syndromes
Highly penetrant LoF variants were identified in TP53 (0.6%) and PTEN (0.2%), associated with Li-Fraumeni syndrome (LFS) and Cowden syndromes, respectively. No mutations were detected in STK11 and CDH1 genes known to cause Peutz-Jeghers and Hereditary Diffuse Gastric Cancer syndromes, respectively.
All individuals carrying TP53 mutations (variant allele frequencies ranged from 46% to 51%) had very young age at diagnosis (range 20–36 years); however, ascertainment bias on age selection should be noted. Among them, 11 BC emerged, seven of which were human epidermal growth factor receptor 2 positive. Surprisingly, only 2/8 initially fulfilled the revised Chompret criteria for TP53 genetic testing,13 followed by two more cases, only after a family relative was diagnosed with an LFS-related cancer; therefore, half of the carriers fulfilled the criteria after later follow-up.
Among three PTEN carriers, only one (F975) fulfilled the testing criteria for Cowden syndrome, due to her clinical features (gastrointestinal hamartomas from young age, thyroid and endometrial cancer diagnosis). Notably, proband F1097, who is a PTEN carrier, also carries a PALB2 deleterious mutation. Available family pedigrees for TP53 and PTEN carriers are summarised in the online supplementary figure 1.
Supplemental material
Moreover, six individuals (6/1382; 0.4%) harboured damaging variants in Lynch-related genes and specifically, three MSH6, two PMS2 and one MLH1 LoF variants were detected. Interestingly, two patients (1178 MLH1 carrier and 1272 MSH6 carrier) had family histories of both breast and gastrointestinal cancers, while patient 2514 (PMS2 carrier) had breast and ovarian cancer and no Lynch-related cancers diagnoses among family relatives.
Individuals carrying two LoF variants
In total, 0.7% (10/1382) of the individuals tested were found to carry two LoF variants in two genes, of which three had combinations of variants both associated with high BC risk, that is, BRCA1 and BRCA2, BRCA1 and PALB2 and PTEN and PALB2 (F543, F330 and F1097, respectively). Four cases combined damaging variants conferring high BC risk with moderate breast or ovarian cancer risks, namely, PALB2 and ATM, BRCA2 and ATM, BRCA2 and RAD51C and PALB2 and SLX4 (F776, F882, F2540 and F2364). Another case (F2070) had a BRCA2 along with a RECQL4 LoF variant, for which little evidence on BC association exists. Single cases involved combinations of ERCC3/FANCC and BRCA2/SDHC LoF variants (F2516 and F1281, respectively).
Interestingly, two patients were also diagnosed with epithelial ovarian cancer (F2070 and F2364). Since most of these gene combinations are either rare or unique, it is rather difficult to investigate whether these individuals present a more severe phenotype, when compared with single mutation carriers, while information on gene mutations can be rather useful for targeted testing and appropriate surveillance on family relatives. All the aforementioned PVs are depicted in blue colour in the online supplementary table 3, while available pedigrees are displayed in the online supplementary figure 2.
Supplemental material
Alleles in genes, other than BRCA1 and BRCA2, with possible founder effect in Greek patients
During this large-scale analysis, a number of PVs were seen recurrently. This is not surprising, since the Greek population is characterised by founder effects, as already reported.3 Four detected variants specifically, PALB2 (c.2257C>T), ATM (c.1215delT), RAD51C (c.706-2A>C) and CHEK2 (c.549G>C) have been encountered in four, apparently non-relative families, each, while CHEK2 c.499G>A has been detected in three, apparently non-relative families. Although these observations suggest possible founder effect(s), additional studies are required to elucidate this.
Association of gene LoF variants with age at primary BC diagnosis, secondary cancer diagnosis and histopathology characteristics
Possible association of age at first and second primary BC diagnosis, ovarian or other cancer and triple-negative breast cancer (TNBC) diagnosis with PVs in BRCA1, BRCA2, PALB2, ATM, CHEK2, TP53 and RAD51C were investigated. The small number of mutation carriers in other genes deterred us from pursuing further analysis.
The analysis results of the are summarised in table 2.
Association of age at primary breast cancer diagnosis, incidence of secondary cancer diagnosis and histopathology with BRCA1, BRCA2, PALB2, ATM, CHEK2, TP53 and RAD51C LoF variants
BRCA1 and TP53 PVs were associated with younger age at first BC diagnosis (mean age 38.74±9.2; p<0.01 and 30.3±6.2; p=0.024, respectively). As expected, BRCA1 and BRCA2 carriers were more likely to develop a second primary BC (p<0.01 and p=0.036, respectively) and to be diagnosed with ovarian cancer (p<0.01 and p=0.013). LoF BRCA1 variants were clearly associated with TNBC diagnosis (p<0.01).
TP53 mutation carriers had a statistically significant higher incidence of second primary BC tumours than non-carriers (p=0.035), while CHEK2 mutation carriers had a significantly higher incidence of other cancer diagnosis, beyond breast or ovarian (p=0.046).
RAD51C deleterious variants seem to be associated with a second BC diagnosis, illustrated by the trend towards significance (p=0.057). Incidence of second primary tumours, other than breast or ovarian, is on the cusp of statistical significance (p=0.063) among TP53 mutation carriers. These results indicate the need of further investigation in larger datasets.
Clinical actionability of LoF variants in other than BRCA1 and BRCA2
Based on the January 2019 National Comprehensive Cancer Network (NCCN) consensus guidelines,14 clinical management and surveillance will be modified for the majority (85/93) of the non-BRCA1/2 mutation carriers (in alphabetical order, ATM, BRIP1, CHEK2, MLH1, MSH6, NBN, PALB2, PMS2, PTEN, RAD51C, RAD51D, TP53, SDHB and SDHC). Herein, breast and ovarian cancer surveillance could be altered in 4.5% (carriers of PALB2, ATM, CHEK2, TP53, PTEN and NBN PVs) and 1.3% (carriers of BRIP1, RAD51C, RAD51D, MLH1 and MSH6 PVs), respectively. Cancer surveillance for other sites (colon, endometrium, abdomen, thyroid) could be modified in 2.96% (carriers of CHEK2, TP53, PTEN, PMS2, MLH1, MSH6, SDHB and SDHC PVs) of the initial cohort. A fundamental aspect of this study was the possibility for targeted testing of the familial mutation in 114 family relatives of the aforementioned mutations carriers, in genes beyond BRCA1/2; of which, 42.9% (49/114) tested positive for the familial PV.
Taken together, 6.1% of the individuals included in this study, along with 49 of their family relatives, would be given the opportunity to adjust their clinical management. This is in addition to BRCA1/2 carriers, who already receive tailored management strategies. Detailed numbers of tested individuals, along with relevant surveillance guidelines per gene are summarised in table 3.
Clinical actionability by site of loss-of-function variants in genes other than BRCA1 and BRCA2
Discussion
This is an original study questioning an extensive number of genes, known to be implicated in cancer predisposition and DNA repair, and their possible association to BC predisposition, in a highly selected cohort of Greek patients with BC. Through this approach, integrated knowledge on BC susceptibility is acquired; from providing a spectrum of variants in known genes, to proposing novel BC candidates, while providing estimates on BC risks.
Among 1382 patients with very young BC diagnosis (<35 years) and/or strong family history, 31.5% harboured LoF variants in at least one gene. Other than BRCA1/2, 4.5% of patients carried variants in genes conferring high and moderate BC risk, that is, CHEK2, ATM, TP53 and PALB2, with detected frequencies being in line with previous studies.15–17
For the first time herein, RAD51C LoF variants show a clear association to BC predisposition, irrespectively of breast histology subtype, while a trend towards secondary BC diagnosis was observed. Damaging RAD51C and RAD51D variants are known to confer elevated ovarian cancer risks, but their association to BC susceptibility is often disputed. RAD51D LoF variants have been associated with moderate BC susceptibility in several studies, even though the number of carriers was limited, while RAD51C damaging variants did not meet statistical significance for association.16 18–20 Quite recently though, an association of both RAD51C and RAD51D PVs to TNBC diagnosis specifically, was reported.21 The association observed in our study can be possibly attributed to the strict patient selection, but also to the relatively large number of RAD51C damaging variants identified, possibly due to a Greek founder effect. If this is further validated, it will be of clinical importance for the appropriate follow-up and management of these individuals.
Primarily, a statistically significant association of CHEK2 missense LoF variants and moderate BC susceptibility, arose. This observation might be due to our population-specific elevated frequency of CHEK2 damaging missense variants, thus allowing statistical associations, while we have been able to evaluate the effect of these missense variants through the implementation of a functional assay.22 On the contrary, when comparing altogether CHEK2 LoF variants, with controls, the association was marginally moderate, and was in compliance with previous reports.16 18 19
We have been able to replicate the established associations of BRCA1 and TP53 LoF variants to early age and second primary BC diagnosis.18 23 Interestingly, second primary tumour diagnosis, beyond breast or ovarian cancer, was found to be associated with the presence of CHEK2 LoF variants. CHEK2 variants have been identified among young individuals diagnosed with colorectal cancer24 and have been previously associated with increased colorectal cancer risk.25 Moreover, prostate26 and gastric cancer risk27 seems to be elevated among CHEK2 carriers. Studies like the current assist in defining these important associations, since although colon surveillance is proposed by NCCN guidelines for CHEK2 carriers, evidence for other cancer-specific risks is limited.14
Mutations in the ‘syndromic’ cancer genes, TP53, PTEN, STK11 and CDH1 were observed at considerably variable frequencies, corresponding to the extreme low end of the prevalence reported in most studies.17 28 Herein, no CDH1 or STK11 damaging alleles were identified, while the prevalence of PTEN and TP53 mutations was 0.2% and 0.6%, respectively, mainly attributed to the prominent syndromic features that are frequently associated with mutations in these genes, allowing direct single gene testing.13
A very important observation that emerged though, involved the weakness of the criteria for LFS genetic testing. In our series, only half of TP53 carriers fulfilled the revised Chompret criteria at diagnosis. As already indicated, testing for LFS based solely on the Chompret criteria will miss a significant number of TP53 mutation carriers,29 30 leading to severe implications for proper patient management, that is, avoidance of radiation due to increased risk for secondary radiation-induced malignancies. It is very likely that a rare, differential or divergent or attenuated LFS subgroup, characterised by early-onset BC, exists and does not fit to the typical LFS phenotype of cancer diagnoses at a very young age, while as recently suggested,30 the LFS phenotypic spectrum will be fully uncovered in the near future. Based on these, LFS testing criteria should be amended to recommend TP53 genetic testing in all female patients with BC diagnosed <35 years.
An important aspect of this study lies within the clinical translation of the produced results, an issue that is on the spotlight since the incorporation of panel testing in clinical setting. Beyond BRCA1/2, the vast majority (91.3%) of the identified carriers, will be offered cancer screening and/or preventive measures, based on current gene-based NCCN guidelines.14 More specifically, a third of the mutation carriers can receive tailored recommendations for multiorgan cancer screening following established guidelines, that is, TP53, PALB2, PTEN, MLH1, MSH6, PMS2, SDHB and SDHC, even in the absence of gene-specific phenotypic characteristics. The benefit of these findings is augmented through the additional testing of family relatives, of which herein, 49 carried the familial mutation and would be therefore given the opportunity to receive more appropriate clinical management.
Noteworthy, 2% of patients in our cohort carried deleterious variants in genes that have been previously suspected to be associated with BC predisposition, while LoF variants in candidate BC genes, either involved in DNA damage repair through the Fanconi anaemia pathway or associated with other known cancer syndromes, were identified in 0.8% of patients in our cohort. Of these, LoF variants in ERRC3 31 and FANCM 32 stand out, since they have been recently associated with BC predisposition. These data, due to the rarity of these variants, should be handled with caution though and their possible association to BC should be interrogated through large case–control studies.
This study is limited to patients with BC of Greek descent, with the cancer risk estimations deriving from comparisons with selected controls from the ExAC and the FLOSSIES data set, which can provide legitimate estimates for the purposes of this study, but do not derive from a balanced case–control study. Another limitation of our study involves the low number of variants in multiple genes, a fact that does not provide the adequate statistical power to draw safe conclusions on whether a LoF variant of a gene can be associated with BC. Moreover, although the gene panel implemented herein is quite extensive, BARD1, PVs of which have been associated with both breast and ovarian cancer predisposition has not been evaluated.
Overall, this study reports a high rate of cancer predisposing variants, followed by calculated risks in known and suspected BC genes, while proposing a number of novel candidates, in a strictly selected cohort of Greek patients with BC. The genetic diversity of the Greek population, simultaneously harbouring ancient founder alleles and characterised by genetic heterogeneity, can assist to BC risk estimation conferred by damaging variants in specific genes, while paving the way for novel BC associations, through the identification of population-specific alleles in additional genes.
Delineating the actual cancer risk associated with specific gene variants will ultimately lead to the development of appropriate clinical management guidelines. As such, the clinical value for both patients with cancer and healthy mutation carriers will be maximised, while moving away the scepticism regarding the clinical validity of multigene panel testing.
References
Footnotes
Contributors FF wrote the manuscript. FF and IK conceived and designed the work and interpreted the data. IK made essential contributions to the manuscript. PA, MSP and AV were involved in the laboratory analyses, collected family pedigrees and corrected the manuscript. CP, EF, CC, IB, ER, DT, VB were involved in patient collection, acquired patient data and corrected the manuscript. ISV and DK were involved in the bioinformatic analyses and interpretation of the results and corrected the manuscript. GF designed multiple aspects of the study and made essential contributions to the manuscript. DY conceived and supervised the study and made essential contributions to the manuscript. All authors provided critical feedback and approved the final version of the manuscript.
Funding This research was co-financed by the European Union (European Social Fund, ESF) and Greek national funds through the Operational Program ‘Education and Lifelong Learning’ of the National Strategic Reference Framework (NSRF) - Research Funding Program of the General Secretariat for Research & Technology (ARISTEIA 39, P. BROCA), which invests in knowledge society through the ESF.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval The study was approved by the Bioethics committee of NCSR ‘Demokritos’ (BCNCSRD-240/EHΔ/11.3, updated on 29 June 2015).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.