Article Text

Abstract
Background Brain arteriovenous malformations (BAVM) represent a congenital anomaly of the cerebral vessels with a prevalence of 10–18/100 000. BAVM is the leading aetiology of intracranial haemorrhage in children. Our objective was to identify gene variants potentially contributing to disease and to better define the molecular aetiology underlying non-syndromic sporadic BAVM.
Methods We performed whole-exome trio sequencing of 100 unrelated families with a clinically uniform BAVM phenotype. Pathogenic variants were then studied in vivo using a transgenic zebrafish model.
Results We identified four pathogenic heterozygous variants in four patients, including one in the established BAVM-related gene, ENG, and three damaging variants in novel candidate genes: PITPNM3, SARS and LEMD3, which we then functionally validated in zebrafish. In addition, eight likely pathogenic heterozygous variants (TIMP3, SCUBE2, MAP4K4, CDH2, IL17RD, PREX2, ZFYVE16 and EGFR) were identified in eight patients, and 16 patients carried one or more variants of uncertain significance. Potential oligogenic inheritance (MAP4K4 with ENG, RASA1 with TIMP3 and SCUBE2 with ENG) was identified in three patients. Regulation of sma- and mad-related proteins (SMADs) (involved in bone morphogenic protein (BMP)/transforming growth factor beta (TGF-β) signalling) and vascular endothelial growth factor (VEGF)/vascular endotheliual growth factor recepter 2 (VEGFR2) binding and activity (affecting the VEGF signalling pathway) were the most significantly affected biological process involved in the pathogenesis of BAVM.
Conclusions Our study highlights the specific role of BMP/TGF-β and VEGF/VEGFR signalling in the aetiology of BAVM and the efficiency of intensive parallel sequencing in the challenging context of genetically heterogeneous paradigm.
- brain arteriovenous malformation (bavm)
- whole exome sequencing
- genetics heterogeneity
- vasculogenesis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
A brain arteriovenous malformation (BAVM) is an anomaly within the cerebral vasculature characterised by high-flow, fragile tangles of dysplastic vessels, forming a nidus composed of feeding arteries and draining veins that shunt blood from the arteries to the veins without an intervening capillary network.1 With an annual detection rate of 1.3 per 100 000 and a prevalence of 10–18 per 100 000, BAVM is the leading cause of intracranial haemorrhage in children and also contributes to adolescent epilepsy.2
While BAVM is usually non-syndromic, it can present in congenital syndromes featuring vascular malformations, such as hereditary haemorrhagic telangiectasia (HHT; OMIM #PS187300) mainly caused by germline, heterozygous loss of function mutations in either ENG,3 ACVRL1,4 BMPR2 5 or SMAD4,6 or in capillary malformation-arteriovenous malformation (CM-AVM; OMIM #608354), which is caused by heterozygous loss of RASA1 activity,7 as well as in Sturge-Weber syndrome (OMIM #185300), which is caused by somatic activating mutations in GNAQ.8 However, potential genetic factors and ‘disease genes’ underlying non-syndromic BAVM remain poorly defined, although recent reports highlight a role for KRAS and mitogen-activated protein kinase (MAPK) signalling in sporadic BAVM formation.9
Zebrafish represent an ideal model organism for studying BAVM due to the transparency of the skull during development, readily available endothelial fluorescent reporter lines that enable high-resolution imaging and the ease of gene knock-down using antisense morpholinos or CRISPR/Cas9 technologies.10 Additionally, the genetic regulators of vascular development and arteriovenous specification are well conserved across zebrafish, mice and humans.11 For example, mutants and morphants of eng and alk1, the zebrafish homologues of the human transforming growth factor beta (TGF-β) receptors ENG and ACVRL1 , are widely used as a BAVM model in molecular and therapeutic studies.12 13
To enhance our understanding of the genetics underlying sporadic BAVM and potential molecular aetiologies, we applied a rare variant approach using whole-exome sequencing (WES) in a cohort of 100 trios composed of probands with uniform diagnosis of BAVM and their phenotypically normal parents. Rare damaging variants were functionally validated using a zebrafish model.
Methods
Cohort collection
Patients diagnosed with BAVM between November 2015 and November 2016 at Beijing Tiantan Hospital, Beijing, China, were consecutively enrolled in our study. Inclusion and exclusion criteria and detailed phenotypic data are provided in the online supplementary methods. We followed up with parents of probands with inherited candidate variants, including pathogenic variants, likely pathogenic variants and variants of uncertain significance (VUS). BAVM-involving phenotypes, such as headache, dizziness and seizure, were rechecked by phone call. An MRI of the brain was taken when possible.
Supplemental material
WES and variant interpretation
WES was performed on DNA extracted from the peripheral blood of 100 trios (probands with phenotypically normal parents). In brief, Illumina paired-end libraries were prepared from DNA samples and subjected to whole-exome capture using the SureSelect Human All Exon V6+UTR r2 core design (91 Mb, Agilent), followed by sequencing on an Illumina HiSeq 4000 platform. In-house developed Peking Union Medical College Hospital Pipeline and variant interpretation methods are provided in the online supplementary methods.
Sanger sequencing
All pathogenic variants, likely pathogenic variants, and VUS were validated by Sanger sequencing. Variant-encoding amplicons were amplified by PCR from genomic DNA obtained from probands and parents and purified using an Axygen AP-GX-50 kit (lot no. 05915KE1) and sequenced by Sanger sequencing on an ABI3730XL instrument.
Zebrafish husbandry and fertilisation
Tg(kdrl.4:mCherry)pku6 transgenic zebrafish, where mCherry expression is driven by a kdrl.4 promoter (an endothelial cell-specific gene), were used.14 Maintenance of adult zebrafish and embryos is described in online supplementary methods.
Morpholino injection and mRNA rescue experiments
Morpholino-modified antisense oligonucleotides (GeneTools, 5 ng for each) were injected into embryos at the 1-cell to 2-cell stage. Two morpholinos (one translation blocking and one splice-site targeting) were used for each gene. In rescue experiments, the splice-site targeting morpholino (5 ng) was coinjected with wild-type/mutant human mRNA (200 pg) for each gene. Morpholino sequences, validation of splice-disrupting morpholinos by reverse transcription (RT)-PCR and synthesis of human mRNA are described in detail within online supplementary methods.
Fluorescence imaging and phenotype evaluation
Fluorescence images were collected at 48hpf by confocal microscopy as Z-series stacks and reconstructed as both a single, two-dimensional maximum intensity projection image and a Z-stack movie. Phenotypic evaluations were performed by two experimenters blinded to the experimental conditions.
Statistics
SPSS Statistics V15.0 software was used for statistical analyses. Statistical significance was defined as p<0.05. Zebrafish phenotype proportion data were analysed using the Student’s t-test.
Results
Cohort enrolment
A total of 123 families of Chinese Han ethnicity with a proband having a presumptive clinical diagnosis of BAVM were consecutively enrolled. After clinical evaluation and imaging examinations, 23 families were excluded (figure 1). This resulted in 100 patients with sporadic, apparently non-syndromic BAVM. Cohort demographics are described in online supplementary table S1. WES was performed on the patients and their phenotypically normal parents. Patients with positive findings (pathogenic variant, likely pathogenic variant and VUS) are described in figure 2 and online supplementary case decriptions.
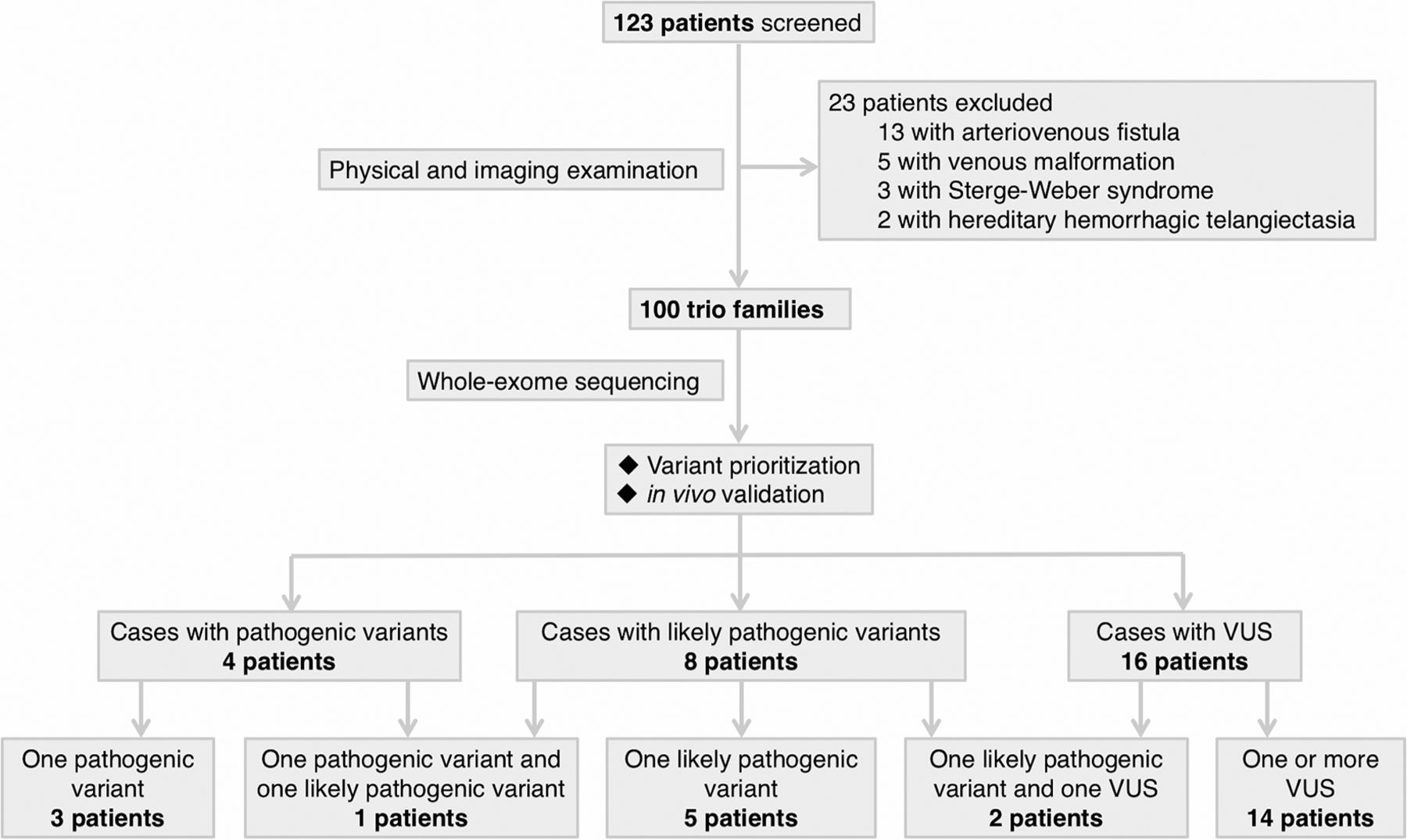
Workflow of patient enrolment and analysis of whole-exome sequencing data. Whole-exome sequencing was conducted in 100 patients with a diagnosis of brain arteriovenous malformation and their normal parents. Variants are prioritised based on: (1) de novo or not; (2) in vivo validation using zebrafish; (3) gene function and expression pattern; (4) frequency in public databases; and (5) recurrence among cohort. We identified 4 pathogenic variants in 4 patients, 8 likely pathogenic variants in 8 patients and 18 VUS in 16 patients. Potential oligogenic models were identified in three patients. VUS, variant of uncertain significance.

Variant information, phenotype and in vivo functional study of PITPNM3, SARS and LEMD3 variants in patient AVM306, AVM464 and AVM334. (A) Results of Sanger sequencing of the de novo variants in PITPNM3, SARS and LEMD3; brain MRI (BMRI) and digital subtraction angiography (DSA) demonstrating the arteriovenous malformation (arrows) in patient AVM306, AVM464 and AVM334. (B) Confocal imaging (dorsal view, anterior to the right) of Tg(kdrl.4:mCherry)pku6 transgenic fish injected with 5 ng of control morpholino or 5 ng of morpholino targeting pitpnm3, sars or lemd3 at 48 hours postfertilisation (hpf). sars and lemd3 morphants were then coinjected with 200 pg of human mRNA. Note the basilar artery (BA), basal communicating artery (BCA), anterior cerebral artery (ACA), metencephalic arteries (MtA), primordial hidbrain channel (PHBC), primordial midbrain channel (PMBC) and posterior connecting segments (PCS). AVMs of BA/PHBC or BCA/PMBC is labelled with ‘#’. (C) Wiring diagrams of BAVM in 48 hpf compared with normal brain vessels. Red wires represent arteries; blue wires represent veins. (D) Significant difference existed between the percentage of embryos displaying BAVM phenotype between pitpnm3 morphants (MO1: 73%, 27/37; MO2: 68%, 26/38) and control embryos (0%) (Student’s t-test, MO1: p=0.00000037; MO2: p=0.0082). Coinjection of pitpnm3 MO2 and pitpnm3 mRNA resulted in reduced percentage of BAVM embryos (17%, 6/35, p=0.00043). Percentage of embryos exhibiting a BAVM-like phenotype between sars morphants (MO1: 83%, 30/36; MO2: 79%, 34/43) and control embryos (0%) was significantly different (Student’s t-test, MO1: p=0.000045; MO2: p=0.00025). BAVM-like phenotype was rescued by human wild-type SARS mRNA (41%, 15/37) but not mutant SARS mRNA (74%, 25/34) (Student’s t-test, p=0.018). A significant difference in the percentage of embryos exhibiting BAVM-like phenotype was detected between the lemd3 morphants (MO1: 82%, 31/38; MO2: 76%, 35/46) and control embryos (0%) (Student’s t-test, MO1: p=0.00094; MO2: p=0.00075). The BAVM-like phenotype was rescued by human wild-type LEMD3 mRNA (24%, 9/38) but not mutant LEMD3 mRNA (66%, 25/38) (Student’s t-test, p=0.0015). Error bars represent one SD, n=3 replicates. A different clutch of embryos was used for each replicate. *P<0.05; **p<0.01. BAVM, brain arteriovenous malformation; MO1, morpholino targeting splice site; MO2, morpholino targeting AUG start codon site; MU, mutant; WT, wild-type.
Spectrum of mutations in genes involved in syndromes including a BAVM phenotype
Because the genetics underlying non-syndromic BAVM have only recently begun to be understood, we first examined the sequences of genes known to cause syndromes associated with AVM, including HHT,3–6 CM-AVM7 and Sturge-Weber syndrome.8 In patient AVM558, a maternally inherited pathogenic heterozygous frameshift variant, c.920dupA (p.Asn307LysfsTer27), was identified in ENG (probability of loss-of-function intolerance15 (pLI)=1) (table 1). We followed up with the mother, who also carried this variant. Brain MRI showed no sign of BAVM, and no HHT-associated phenotypes, such as epistaxis, telangiectasis or other visceral involvement were identified in either the patient and the mother. ENG encodes Endoglin, a transmembrane accessory receptor essential for normal TGF-β signalling in endothelial cells.16 ENG haploinsufficiency causes HHT (OMIM #PS187300), which clinically manifests as pulmonary AVM in 41% of patients.17 ENG +/− mice develop cerebral AVM with a penetrance of 30%,18 which could explain the incomplete penetrance of BAVM in this family.
Pathogenic and likely pathogenic variants in known and candidate genes associated with BAVM
Additional VUS with allele counts in ExAC, but with a minor allele frequency <0.01, were identified in ENG, ACVRL1 and BMPR2 (two other HHT genes) and RASA1 (table 2).
Variants of uncertain significance in known and candidate BAVM genes
Pathogenic de novo variants identified in PITPNM3, SARS and LEMD3
A total of 85 rare, functional (missense, nonsense, splice site and insertion or deletion) de novo variants were identified, of which one heterozygous predicted intolerable truncating variant c.274C>T (p.Arg92Ter) in PITPNM3 (pLI=1) was identified in AVM306 (table 1). PITPNM3 promotes the invasion of breast cancer cells through PI3K/Akt pathway, which regulates angiogenesis both in tumour and normal tissues.19 To examine the effect of PITPNM3 depletion in vivo, we designed two non-overlapping morpholino oligonucleotides (MOs) to knockdown pitpnm3 in zebrafish. pitpnm3 MO1 (a splice-blocking MO targeting the exon 5: intron 5 boundary) or MO2 (a translation blocking MO targeting the AUG ‘start’ codon) were each injected into embryos containing a panendothelial fluorescent reporter allele, Tg(kdrl.4:mCherry)pku6 .14 Confocal microscopy revealed that embryos injected with either pitpnm3 MO1 (73%) or MO2 (68%) (but not control morphant or uninjected embryos) recapitulated a range of human BAVM phenotypes characterised by: (A) dilation and deformation of the basal communicating artery (BCA) and posterior connecting segments; (B) direct shunting between basilar artery/primordial hindbrain channel or BCA/primordial midbrain channel (PMBC); (C) fusion between the BCA and PMBC; and (D) dysplasia of the anterior cerebral arteries (figure 2B, online supplementary videos 1–3). These results are consistent with other zebrafish models of BAVM12 13 (figure 2C). The BAVM phenotype can be rescued by coinjection of morpholino and pitpnm3 mRNA (figure 2B, online supplementary video 4), validating the specificity of the targeted knockdown. Most of the pitpnm3 morphants featuring BAVM also exhibited cranial and pericardial oedema (online supplementary figure S1), likely due to abnormal circulation, as most of the blood flowed through a limited number of dilated cerebral vessels, with little circulation through the trunk and tail.20 Therefore, haploinsufficiency of PITPNM3 most likely contributed to the BAVM in AVM306.
Supplemental material
We also identified a de novo missense variant in both SARS and LEMD3 involving dominant loss of function (LoF) alleles that were predicted to cause BAVM. A de novo heterozygous missense variant in SARS, c.971T>C (p.Ile324Thr) (pLI=1) (table 1), was identified in patient AVM464. Mutation or MO knockdown of sars in zebrafish reportedly causes ectopic branching of brain and segmental vessels,21 22 a phenotype highly reminiscent of AVM. sars is expressed in the early somites, brain and notochord, all tissues known to express vegfa and loss of sars upregulates vegfa transcript levels.21 22 Under the hypothesis that this de novo SARS variant contributes to the BAVM phenotype via a LoF mechanism, we used one previously validated morpholino targeting the exon8-intron8 splice-site,21 22 as well as a non-overlapping morpholino targeting the start codon of sars. Both sars morphants exhibited a BAVM-like phenotype (MO1=83%, MO2=79%) similar to that of the pitpnm3 morphants described above (figure 2B, online supplementary videos 5 and 6). sars morphants also exhibited cranial and pericardial oedema (online supplementary figure S1) recapitulating previously published studies involving sars loss of function mutants.21 22
To further determine the function of the c.971T>C (p.Ile324Thr) SARS variant, sars morphants were coinjected with WT and c.971T>C mutant human mRNA. Intriguingly, coinjection of human SARS WT mRNA, but not the SARS mutant [c.971T>C] mRNA, rescued the BAVM-like phenotype in sars morphants (figure 2B, online supplementary videos 7-8), as indicated by a decreased proportion of embryos with brain vessel abnormalities (figure 2D), thus identifying c.971T>C (p.Ile324Thr) in SARS as a pathogenic variant. sars encodes a seryl-transfer RNA synthetase, but this enzymatic function is dispensable for its role in vascular development.21 22 The mutation we identified falls near the L-serine binding site (p.325Glu) of SARS,23 but how it affects SARS non-canonical activity in vascular development remains to be determined (as does whether this mutation affects Sars seryl-tRNA synthetase activity).
The de novo heterozygous missense variant c.2636C>G (p.Thr879Ser) in LEMD3 (pLI=1) was identified in patient AVM334 (table 1). While expression data are not available for lemd3 in zebrafish, previous studies show that it is expressed within the developing vasculature of the early embryonic mouse.24 Xenopus and human LEMD3 (previously known as MAN1) localises to the inner nuclear envelope and interacts with bone morphogenic protein (BMP)-responsive SMADs (Smad1, Smad5 and Smad8) through its c-terminus.24 Structure–function experiments in Xenopus demonstrated that LEMD3 antagonises BMP through this carboxy-terminal region.25 Arteriovenous malformations were not reported in Lemd3−/+ or Lemd3 null mutants, but severe cardiac oedema, compromised embryonic turning, altered yolk sac vessel remodelling and embryonic lethality at ~E11.5 were noted in the homozygous mutants, as were left–right patterning defects.26 lemd3 morphants (generated by two different, non-overlapping morpholinos) exhibited a highly penetrant BAVM-like phenotype (figure 2B, online supplementary videos 9-10) (MO1=82%, MO2=76%), confirming the role of zebrafish lemd3 in vascular development. In addition to brain vessel malformations and oedema at multiple sites, lemd3 morphants also exhibited truncated body length, which is probably due to defects in axis formation and reduced anterior neuroectoderm (as occurs in Xenopus following lemd3 knockdown).25 Lemd3 is composed of four domains: an amino-terminal LEM domain, two transmembrane domains and a c-terminal RNA-recognition motif (RRM). The predicted amino acid change affects a residue within this RRM motif. Injection of human LEMD3 WT mRNA, but not LEMD3 mutant [c.2636C>G] mRNA (figure 2B, online supplementary videos 11 and 12), rescued the BAVM-like phenotype in lemd3 morphants (figure 2D), suggesting that the c.2636C>G (p.Thr879Ser) mutation is pathogenic.
The efficiency of the splice MOs for any of the targets (ie, pitpnm3, lemd3 and sars), as measured by aberrant mRNA transcript production, did not reach 100% efficiency, likely due to a combination inefficient targeting (that comes with most antisense oligo technologies), suboptimal seed sequence selection during target design and occasional suboptimal delivery (as can happen during manual injection of hundreds of embryos that are then pooled for subsequent RT-PCR analysis). However, in all cases, two separate, non-overlapping MOs generated identical results that were rescued by adding back WT mRNA, and in the case of sars, the MO recapitulated the established mutant phenotype.21 22
Likely pathogenic de novo variants in candidate pathways
The genes containing the remaining de novo variants were screened to identify those involved in the primary pathways of angiogenesis (figure 3). Seven likely pathogenic de novo missense variants were identified in seven families (table 1).

{kind=link}
{kind=link}
{kind=link}
Major angiogenesis pathways associated with genes identified in the present study. Major components and regulators of the BMP/TGF-β, VEGF, PI3K/Akt and Notch signalling pathways are presented in black. Genes in red harbour pathogenic variants; genes in orange harbour likely pathogenic variants; genes in yellow harbour variants of uncertain significance. Solid arrows indicate activation; solid bars indicate inhibition; dashed arrows indicate general interactions. TGF-β, transforming growth factor beta.
In patient AVM028, the de novo heterozygous missense variant c.311T>C (p.Leu104Pro), in the functional inhibition of zinc metalloproteinases (NTR) domain, was identified in TIMP3 (table 1), which encodes a tissue metalloproteinase inhibitor. TIMP3 inhibits VEGF-mediated angiogenesis by blocking VEGF/VEGFR2 binding (figure 3), a function considered independent of metalloproteinase inhibition and unique to TIMP3 compared with other known TIMPs.27
In patient AVM359, the de novo heterozygous missense variant c.1592G>A (p.Cys531Tyr) was identified in SCUBE2 (table 1), which encodes a membrane-associated multidomain protein. The variant is predicted to affect a conserved site (SIFT=0, PolyPhen2=1, GERP++=5.68, CADD=24.6). SCUBE2 forms a complex with VEGF and VEGFR2 and acts as a coreceptor to enhance VEGF/VEGFR2 binding, thus stimulating VEGF signalling28 (figure 3). The c.1592G>A (p.Cys531Tyr) SCUBE2 variant could induce BAVMs via a gain-of-function mechanism, though confirmation will require further functional studies.
In patient AVM558, the de novo heterozygous missense variant c.1694G>A (p.Arg565Gln) was identified in MAP4K4 (table 1), which encodes a kinase responsible for phosphorylation of residue T312 within SMAD1, blocking SMAD1 activity in BMP/TGF-β signalling (figure 3).29 Loss of MAP4K4 leads to impaired angiogenesis in vitro and in vivo.30
In patient AVM206, the de novo heterozygous missense variant c.2075A>G (p.Asn692Ser) was identified in CDH2 (table 1), which encodes N-cadherin, an integral mediator of cell–cell interactions.31 N-cadherin mediates brain angiogenesis by stabilising angiogenic capillaries, possibly by enhancing the interaction between pericytes and endothelial cells.31 At the molecular level, N-cadherin mediates cell–cell adhesion by regulating PI3K/Akt signalling (figure 3).32
In patient AVM467, the de novo heterozygous missense variant c.676G>A (p.Gly226Ser) was identified in IL17RD (table 1). IL17RD is highly expressed in vessel endothelial cells and vascularised organs, where it inhibits fibroblast growth factor (FGF) and plays critical roles in endothelial cell proliferation and angiogenesis.33 In contrast to FGF inhibition, overexpression of IL17RD attenuates the degradation of epidermal growth factor recepter (EGFR) and enhances downstream MAPK signalling (figure 3).34
In patient AVM457, a de novo heterozygous missense variant c.3355G>A (p.Ala1119Thr) with a robust deleterious damaging predictions (SIFT=0.1, PolyPhen2=0.99, GERP++=4.33, CADD=29.3) was identified in PREX2 (table 1). PREX2 activates PI3K signalling via inhibition of phosphatase and tensin homolog (PTEN),35 and both germline and mosaic PTEN variants are associated with AVMs.36
In patient AVM427, the de novo heterozygous missense variant c.3442G>T (p.Asp1148Tyr) was identified in ZFYVE16 (table 1), which encodes an endosomal protein also known as endofin. ZFYVE16 is an SMAD anchor that facilitates SMAD1 phosphorylation, thus activating BMP signalling.37 In addition to Smad1-mediated BMP signalling, ZFYVE16 also interacts with Smad4 to mediate Smad2–Smad4 complex formation and facilitate TGF-β signalling,38 indicating a regulatory role in BMP/TGF-β signalling (figure 3).
Other potential dominant genes with incomplete penetrance
We also examined other inherited dominant pathogenic variants potentially involving LoF. Evidence of involvement in the pathogenesis of AVM was found in patient AVM312, who carried a paternally inherited heterozygous nonsense variant, c.1891G>T (p.Glu631Ter), in EGFR (table 1). Oncogenic EGFR stimulates angiogenesis via the VEGF pathway.39 As a truncated germline EGFR variant has not been reported in humans, c.1891G>T (p.Glu631Ter) in patient AVM312 was classified as likely pathogenic and EGFR as a candidate gene due to the vital role of EGFR in EGF and VEGF signalling.40
Recurrent biallelic damaging variants
To assess the possibility of a recessive mode of inheritance, we investigated all homozygous and compound heterozygous variants with either a recurrent or LoF allele. Compound heterozygous variants in DSCAML1, DSCAM and PTPN13 were retained.
In two unrelated patients, AVM106 and AVM285, identical compound heterozygous variants were identified in DSCAML1: c.5783G>A (p.Arg1928His) and c.4574G>A (p.Arg1525His), each inherited from heterozygous carrier parents (table 2). Both variants were reported in ExAC with an allele frequency <0.001, and they were predicted in silico to be highly deleterious (GERP++>4 and CADD>30 for both). In patient AVM226, we identified the compound heterozygous variants c.3775G>A (p.Val1259Ile) and c.2966A>T (p.Gln989Leu) in DSCAM (table 2). DSCAML1 and DSCAM have similar neurodevelopmental functions and are essential for self-avoidance in the developing mouse retina.41
In patient AVM144, the compound heterozygous variants c.116–1G>A and c.1000T>A (p.Ser334Thr) were identified in PTPN13 (table 2).
Potential oligogenic inheritance
Variants in more than one gene (at least one likely pathogenic variant) with differing inheritance origin were identified in three patients (figure 1). In patient AVM558, a pathogenic heterozygous variant c.920dupA (p.Asn307LysfsTer27) inherited from the mother was identified in ENG. Another de novo novel heterozygous missense variant, c.1694G>A (p.Arg565Gln), was identified in MAP4K4 (online supplementary table S2), which encodes the kinase responsible for phosphorylation of residue T312 in SMAD1 to block its activity in BMP/TGF-β signalling.29 This de novo variant may modify the effect of the truncating variant in ENG by repressing BMP/TGF-β signalling.
In patient AVM359, one heterozygous VUS (c.589C>T [p.Arg197Trp]) in ENG inherited from the mother and one likely pathogenic de novo heterozygous variant (c.1592G>A [p.Cys531Tyr]) in SCUBE2 were identified (online supplementary table S2). SCUBE2 functions as a coreceptor that enhances VEGF/VEGFR2 binding to stimulate VEGF signalling.28 In this case, both the TGF-β and VEGF signalling pathways could be affected, potentially causing a more severe downstream effect than would occur with variants in only one of the pathways, with the mutations synergising to lead to BAVM.
In patient AVM028, one novel heterozygous VUS (c.2207A>G [p.His736Arg]) in RASA1 inherited from the father and one likely pathogenic de novo novel heterozygous variant (c.311T>C [p.Leu104Pro]) in TIMP3 were identified (online supplementary table S2). While TIMP3 blocks VEGF/VEGFR2 signalling,27 RASA1 modulates differentiation and proliferation of blood vessel endothelial cells downstream of VEGF (figure 3).7 Therefore, the inherited RASA1 variant and de novo TIMP3 variant could contribute to BAVM via additive effects on the same pathway.
To more completely elucidate details of oligogenic pathogenesis in BAVM, both inherited heterozygous and de novo variants must be carefully examined. In particular, for prenatal genetic counselling, both parental and prenatal DNA should be sequenced to better evaluate the risk of BAVM.
Discussion
We used a WES genomic approach to identify potential contributory genes and investigate the genetics underlying BAVM in a cohort of 100 sporadic trios. Four pathogenic and eight likely pathogenic variants were identified in our cohort, but no significant recurrence of causal variants was observed, suggesting a heterogeneous genetic predisposition to BAVM.
BAVM could be caused by variants in any one of multiple genes, especially when one considers that the BAVM phenotype presents in multiple other vascular syndromes caused by a spectrum of mutations (eg, HHT or CM-AVM).3 4 7 Another factor suggestive of genetic heterogeneity is the variable phenotype among our BAVM patients, particularly the age of onset (which ranged from 3 to 32 years of age (online supplementary table S1)), suggesting that variants in different genes potentially affect different stages of vascular morphogenesis, probably through disrupting distinct biological pathways. The spectrum of both size and the location of the BAVM nidus (see online supplementary case descriptions) also suggests that different genetic mechanisms have a unique effect on brain vessels.
Although genetic heterogeneity has hindered the identification of major BAVM-associated genes, functional variants in genes involved in related biological pathways can be regarded as powerful evidence that enhances our understanding of the pathogenesis of BAVM.
Major components of BMP/TGF-β signalling, including ENG,3 ACVRL1 4 and SMAD4,6 are associated with HHT, which can manifest with BAVM. Novel genes identified in our cohort (LEMD3 and MAP4K4) antagonise BMP/TGF-β signalling.25 29 Interestingly, the TGF-β antagonist losartan attenuates the AVM phenotype in alk1 knockdown zebrafish,13 probably by inhibiting angiotensin receptors.42 Considered together with our human and zebrafish results, we hypothesise that in vivo negative regulators of TGF-β signalling, such as LEMD3 and MAP4K4, are critical for fine tuning the signalling pathway and normalising the function and patterning of the cerebrovasculature. LEMD3 diminishes BMP4-associated upregulation of Smad6 and Smad7 expression in vitro.43 MAP4K4 phosphorylates SMAD1 at residue T312, blocking its activity in regulating BMP signalling (figure 3).29 Another SMAD-interacting gene in which a likely pathogenic variant was identified, ZFYVE16, encodes a SMAD anchor that binds to SMAD to activate BMP signalling.37 ZFYVE16 also interacts with SMAD4 by mediating SMAD2–SMAD4 complex formation, thus facilitating TGF-β signalling.38 While ZFYVE16, unlike LEMD3 and MAP4K4, does not function as a BMP/TGF-β antagonist, ZFYVE16 variants could exert similar effects (eg, dominant negative alleles and so on). Thus, BMP signalling and key regulators of the pathway appear to play critical roles in maintaining cerebrovascular homeostasis.
While activating mutations in VEGF signalling have not been linked to BAVM pathogenesis in humans, ECs isolated from BAVMs display aberrant angiogenic features, including increased migration and endothelial cell turnover,44 as well as poor perivascular coverage.45 VEGF expression is also elevated in human BAVM tissue.46 Additionally, while adult loss of Alk1 (HHT2) or Eng (HHT1) in adult mice is well tolerated, exogenous addition of VEGF (or stimulation of angiogenesis through cranial wounding) robustly produces BAVM in these loss of function settings.47 Critically, inhibition of VEGF signalling in experimental models can prevent BAVM formation.48 Embryonic deficiency in mice of either Vegf, or its key receptors Vegfr2 (Kdrl/Flk) and Vegfr1 (Flt1) disrupts vasculogenesis, as the major axial vessels of the dorsal aortae and cardinal veins fail to form.49 In our patient cohort, a de novo heterozygous missense variant in SARS was identified as causing BAVM in a LoF manner. Previous zebrafish studies reported significantly increased levels of vegfa mRNA in sars mutants, explained by a mechanism, whereby Sars acts as a repressor of vegfa transcription in a non-canonical/tRNA synthetase independent manner.21 22
SCUBE2 and TIMP3 were each found to harbour one likely pathogenic de novo variant, and both affect the binding of VEGF to VEGFR2 (figure 3).27 28 SCUBE2 forms a complex with VEGF and VEGFR2, acting as a coreceptor that enhances VEGF/VEGFR2 binding to stimulate VEGF signalling.28 By contrast, TIMP3 inhibits VEGF-mediated angiogenesis by blocking VEGF/VEGFR2 binding.27 Compared with VEGFR1 and VEGFR3, which primarily function in growth factor release and morphogenesis of lymphatic vessels, VEGFR2 functions primarily in angiogenesis.50 Although SCUBE2 and TIMP3 appear to have an opposing effect on VEGF/VEGFR2 binding, missense variants in these two genes could have a syntrophic effect, though this requires further investigation.
Genetic heterogeneity facilitates the identification and characterisation of biological processes and pathways underlying complex diseases such as BAVM. Pathogenic and likely pathogenic variants identified in unrelated cases provided biological and epidemiological evidence supporting a causal role for the BMP/TGF-β and VEGF pathways in BAVM. Genes associated with regulation of SMADs (in BMP/TGF-β signalling) and VEGF/VEGFR2 binding (in VEGF signalling) are high-priority candidates for further functional studies, genetic screening and targeted interventions.
Due to technical difficulties in acquiring BAVM tissue specimens, we were unable to study tissues for somatic KRAS variants, which have been recently identified in a substantial proportion of BAVM cases.9 However, our study could provide complimentary evidence for those patients whose pathogenesis is not explained by KRAS variants, by showing that besides somatic mutation, germline de novo mutations also contribute to the pathogenesis of BAVM.
In conclusion, we identified four pathogenic variants in both a known gene (ENG) and several novel genes (PITPNM3, SARS and LEMD3) in four BAVM patients. We also identified 8 likely pathogenic variants in 8 patients and 18 VUS in 16 patients. Our results suggest that a substantial proportion of BAVM cases are caused by individual rare pathogenic variants that disrupt the function of genes involved in critical angiogenesis pathways, including BMP/TGF-β and VEGF. In particular, genes regulating biological processes such as SMAD activity (in BMP/TGF-β signalling) and VEGF/VEGFR2 binding (in VEGF signalling) harboured clusters of pathogenic and likely pathogenic variants. We also identified potential oligogenic variants in three patients, each of which carried suspicious inherited and de novo variants, indicating a novel pathogenesis model for BAVM and suggesting the necessity of both prenatal and parental DNA screening. Our data emphasise the power of intensive parallel sequencing in the challenging context of genetic heterogeneity and identified critical biological processes in the pathogenesis of BAVM that warrant further research and clinical attention.
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Acknowledgments
We appreciate all of the patients, their families and clinical research coordinators, including physicians who participated in this project. We would like to thank GeneSeeq Inc. for whole-exome sequencing technical support. We would also like to thank Lianyan Li from Peking University for her kind assistance in zebrafish experiments.
References
Footnotes
KW and SZ contributed equally.
Contributors NW, XY and PL conceived of the project and designed the study. KW, QZ, XD, Jian L, YZ and XY enrolled the cohort. SeZ, BowL, YL, JiaqL, JiacL, WC, XS, XL, YN, BoL and JC conducted the experiments. ZB, YS, JW and XT provided technical support for zebrafish experiments. SeZ, KW, QZ, YW, PL and NW analysed the data. XS, YW and ZY conducted the bioinformatic analyses. RD, CC and YM assisted with data interpretation and revised the manuscript. ZW, JRL, JEP, BZ and ShuyangZ assisted with study organisation and manuscript revision. SeZ, JDW, NW, KW and PL wrote the manuscript.
Funding National Natural Science Foundation of China (81671139 and 81220108007 to XY, and 81501852 to NW), Beijing Nova Program (Z161100004916123 to NW), National Key Research and Development Plan of China (2016YFC1300800 and 2016YFC090150 to XY, 2016YFC0901501 to ShZ), 2016 Milstein Medical Asian American Partnership Foundation Fellowship Award in Translational Medicine (to NW), CAMS Initiative Fund for Medical Sciences (2016-I2M-3-003 to NW), Beijing Natural Science Foundation (7172175 to NW), Central Level Public Interest Program for Scientific Research Institute (2016ZX310177 to NW), PUMC Youth Fund & the Fundamental Research Funds for the Central Universities (3332016006 to NW), Distinguished Youth Foundation of Peking Union Medical College Hospital (JQ201506 to NW), the Beijing Nova Program Interdisciplinary Collaborative Project (xxjc201717 to NW), the American Heart Association (16GRNT31330023 to JW), the National Key Research and Development Program of China, Stem Cell and Translational Research (2016YFA0100500 to XT), the National Institute of Neurological Disorders and Stroke (NINDS R01N058529 to JRL), National Human Genome Research Institute/National Heart, Lung, and Blood Institute (NHGRI/NHLBI UM1 HG006542) and the National Human Genome Research Institute (NHGRI K08 HG008986 to JEP).
Competing interests JRL has stock ownership in 23andMe and Lasergen, is a paid consultant for Regeneron Pharmaceuticals and is a coinventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from the chromosomal microarray analysis and clinical exome sequencing offered in the Baylor Genetics Laboratory (http://bmgl.com).
Patient consent Obtained.
Ethics approval This study was approved by the ethics committee of Beijing Tiantan Hospital under KYSB2016-060. Zebrafish experiments were approved by the Institutional Animal Care and Use Committee of Peking University under reference LSC-ZhangB-1.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.