Article Text

Abstract
Background Chimeric antigen receptor T (CAR-T) cells engineered with lentiviral and retroviral vectors have been successfully applied to treat patients with B cell malignancy. However, viral integration in T cells has the potential risk of mutagenesis, and viral vector production demands effort and is costly. Using non-integrative episomal vector such as minicircle vector to generate integration-free CAR-T cells is an attractive option.
Methods and results We established a novel method to generate minicircle vector within a few hours using simple molecular biology techniques. Since no bacteria is involved, we named these vectors bacteria-free (BF) minicircle. In comparison with plasmids, BF minicircle vector enabled higher transgene expression and improved cell viability in human cell line, stem cells and primary T cells. Using BF minicircle vector, we generated integration-free CAR-T cells, which eliminated cancer cells efficiently both in vitro and in vivo.
Conclusion BF minicircle vector will be useful in basic research as well as in clinical applications such as CAR-T and gene therapy. Although the transgene expression of minicircle vector lasts apparently shorter than that of insertional lentivirus, multiple rounds of BF minicircle CAR-T cell infusion could eliminate cancer cells efficiently. On the other hand, a relatively shorter CAR-T cell persistence provides an opportunity to avoid serious side effects such as cytokine storm or on-target off-tumour toxicity.
- bacteria-free minicircle vector
- integration free car-t cells
- cell viability
- human Cd34+ Hscs
- human es cells
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See:http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- bacteria-free minicircle vector
- integration free car-t cells
- cell viability
- human Cd34+ Hscs
- human es cells
Introduction
Chimeric antigen receptor T (CAR-T) cell therapy is one of the most promising treatments for cancer. In 2017, two CAR-T cell products were approved by the Food and Drug Administration (FDA) for the treatment of acute lymphoblastic leukaemia and advanced lymphomas, respectively.1 Currently, CAR-T cells in majority of the studies, including two FDA-approved products, are generated using lentiviral or retroviral vectors.1 2 Viral integration in T cells has the potential risk of mutagenesis, and the effort and cost of viral vector production and regulatory demands associated with clinical use make this virus-based treatment costly, therefore limiting its broad applications.3–5
Alternatively, non-integrative vectors are attractive options. A high level of transgene expression could be achieved shortly after DNA plasmid delivery into the target cells. However, the expression falls rapidly to a low level within a few days even if the DNA vectors are retained in these cells. It has been reported that bacterial DNA linked to a mammalian expression cassette results in transcriptional silencing of episomal transgene.6 7 To address this issue, minicircle DNA vector devoid of bacterial backbone was developed.6 8 9 Compared with bacterial plasmids, minicircle episomal DNA vectors have more persistent and higher transgene expression in vivo,8 10 which make them attractive tools for gene therapy. Previously, different methods have been developed to produce minicircle vectors using specific producer plasmids and genetically modified bacterial strains, which usually take several days to finish.9 In addition, producing vectors using bacteria could lead to endotoxin contamination.11
In this study, we established a novel method to produce minicircle vector within a few hours using simple molecular biology techniques, without using any bacteria strain. We name this vector bacteria-free (BF) minicircle. Compared with plasmids, BF minicircle vector enabled higher transgene expression and better cell viability in cell line, stem cells and primary T cells. In addition, we generated integration-free CAR-T cells using BF minicircle vector, and they eliminated cancer cells efficiently both in vitro and in vivo, with an efficacy comparable with CAR-T cells engineered with lentiviral vector.
Materials and methods
Production of BF minicircle vector
To amplify target transgene, we designed 96 pairs of primers. The 5’ end of each oligo contains BbsI site followed by 6 bp unique sequences. The PCR products digested by BbsI will have 4 bp single strand overhang at both ends. The total possible combination of these 4 bp overhang is 256 (44), and since the overhang on one end of each PCR product needs to be compatible with that of the other end, the number of possible unique overhang pairs is 128. Ninety-six pairs of primers were randomly selected from these 128 combinations, and the sequences of the primers used in this experiment are shown in online supplementary table S1.
Supplemental material
Using these 96 pairs of primers, the target fragments (EF1a-019-2A-eGFP/CMV eGFP) were amplified from plasmids (Takara, PrimeSTAR HS DNA Polymerase, Cat: #R010B) under the following conditions: 95°C for 5 min; 35 × (95°C for 30 s, 58°C for 30 s, 68°C for 10–40 s); 68°C for 2 min; and hold at 4°C.
PCR products were pooled and purified using Qiagen QIAquick PCR Purification Kit (Cat No/ID: 28106). Restriction endonuclease BbsI was used to digest the PCR product (New England, Cat: #R0539L). After purification (QIAquick PCR Purification Kit), the digested DNA fragments were ligated using T4 ligase (New England, Cat: #M0202L) at 16°C for 2 hours, followed by T5 exonuclease (New England, Cat: #M0363L) treatment at 37°C for 2 hours. The BF minicircle vectors were collected after a final round of DNA purification.
Cell lines
K562 (erythroleukaemia cell line) and Raji (Burkitt’s lymphoma cell line) were purchased from American Type Culture Collection (ATCC). Raji-ffluc for bioluminescent imaging and K562-CD19 cells were generated as previously described.12 All above cell lines were grown under standard conditions in RPMI 1640 medium (Thermo Fisher Scientific). Lentiviral producer cell lines 293T (ATCC-CRL3216) were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific). All mediums were supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin and streptomycin, 2 mM L-glutamine, and 1 mM sodium pyruvate. All cell lines were grown at 37°C in a 5% CO2 atmosphere.
Primary human fresh umbilical cord blood-derived T cells
Umbilical cord blood (UCB) units were obtained from Beijing Cord Blood Bank (Beijing, China) with informed consent from healthy volunteer donors. After Histopaque-1077 (Sigma-Aldrich) gradient separation, mononuclear cells were collected and T cells were isolated using the EasySep Human T Cell Enrichment Kit (Stemcell Technologies). T cells were stimulated with anti-CD3/CD28 Dynabeads (Thermo Fisher Scientific) at a bead to T cell ratio of 1:1 and cultured in X-Vivo 15 medium (Lonza), supplemented with 5% (v/v) heat-inactivated fetal bovine serum, 2 mM L-glutamine and 1 mM sodium pyruvate in the presence of 300 IU/mL recombinant human interleukin (IL)-2 (all from Thermo Fisher Scientific).
Primary human UCB-derived CD34+ haematopoietic stem cells and H9 cells
UCB units were obtained from Beijing Cord Blood Bank with informed consent from healthy volunteer donors. After Histopaque-1077 (Sigma-Aldrich) gradient separation, mononuclear cells were collected and CD34+ haematopoietic stem cells (HSCs) were isolated using the EasySep Human Cord Blood CD34 Positive Selection Kit (Stemcell Technologies, #17896). After selection, CD34+ cells were cultured in StemSpan H3000 (Stemcell Technologies, #09800) added with expansion supplement (Stemcell Technologies, #02691).
Electroporation of K562, CD34+ HSCs and H9 cells
2×105 K562 cells were electroporated with 3 µg PCR-eGFP or mini-eGFP or plasmid-eGFP by 4D-Nucleofector System N (Lonza) using the Amaxa SF Cell Line 4D-Nucleofector X Kit S (V4XC-2032) according to the manufacturer’s instructions. The FF-120 program was used.
5×104 CD34+ HSCs were electroporated with 1 µg mini-eGFP or plasmid-eGFP by 4D-Nucleofector System N (Lonza) using the P3 Primary Cell 4D-Nucleofector X Kit (V4XP-3024, Lonza) according to the manufacturer’s instructions. The EO-100 program was used.
4×105 H9 embryonic stem cells (ESCs) were electroporated with 1 μg mini-eGFP or plasmid-eGFP by 4D-Nucleofector System N (Lonza) using the P3 Primary Cell 4D-Nucleofector X Kit (V4XP-3024, Lonza) according to the manufacturer’s instructions. The CB-150 program was used.
After electroporation, cells were resuspended with prewarmed cell medium and transferred into plate and incubated at 37°C in 5% CO2. Cell viability and transfection efficiency were evaluated 2 days after electroporation by cell counting and fluorescence-activated cell sorting (FACS) analysis, respectively.
Generation of lenti-019-CAR-T cells
Freshly purified primary T cells were activated for 1 day and then transduced with lentiviral vectors harbouring a second-generation CD19 CAR. The structure of the CAR was previously described.12 The infection efficiency was evaluated 2 days after transduction by FACS analysis.
Generation of mini-019-CAR-T cells
T cells were activated for 3 days and then 1×106 cells were electroporated with 3 µg mini-019-CAR DNA by 4D-Nucleofector System N (Lonza) using the P3 Primary Cell 4D-Nucleofector X Kit (V4XP-3024, Lonza) according to the manufacturer’s instructions. The EO-115 program was used. After electroporation, cells were resuspended in 1 mL prewarmed T cell medium and transferred into 12-well cell plate and incubated at 37°C in 5% CO2. Cell viability and transfection efficiency were evaluated 2 days after electroporation by cell counting and FACS analysis, respectively.
Luciferase-based CTL assay
The cytotoxicity of CAR-T cell was assessed according to luciferase-based Cytotoxic T Lymphocyte (CTL) assay described previously.13 K562-luc and K562-CD19-luc cell lines were generated by transducing K562 and K562-CD19 cells with lentivirus expressing luciferase. Briefly, K562-luc and K562-CD19-luc cells were resuspended at 1×105 cells/mL in 100 µL RPMI 1640 medium and mixed with different ratios of effect cells (eg, 10:1, 5:1 and so on), with a total volume of 200 µL. After incubation at 37°C in 5% CO2 for 16 hours, 10 µL of substrate was added, and the luminescence was measured 5 min later. The results are reported as per cent killing based on the luciferase activity in the wells with tumour cells but no T cells (% killing=100 – ((RLU from well with effector and target cell coculture) / (relative light unit (RLU) from well with target cells)×100)).
Cytokine ELISA
Cytokine release assays were performed by coculture of effector cells (T, mini-019-CAR-T) with target tumour cells (K562-CD19; K562) at a 1:1 ratio (104 cells each) per well in duplicate in 96-well plates in a final volume of 200 µL complete RPMI 1640 medium. After 24 hours, supernatants were assayed for production of IL-2 and interferon (IFN)-γ using ELISA Kit (BioLegend).
Murine xenograft studies
For establishing the Raji-ffluc tumour model, NOD-Prkdcscid Il2rgnull (NPG) mice 6–12 weeks old (VITALSTAR, Beijing, China) were injected with 2×105 Raji-ffluc cells via intraperitoneal injection on day 0. Three days after injection, tumour engraftment was evaluated by serial biophotonic imaging using the NightOWL LB983 In Vivo Imaging System (Berthold Technologies). Mice with comparable tumour loads were segregated into different treatment groups and received different treatments: phosphate-buffered saline (PBS) group, T cell group, lenti-019-CAR-T cell group and mini-019-CAR-T cell group. Cells were administered at a dose of 5×106 cells/mouse via intraperitoneal injection. The tumour loads were evaluated 6 days after first treatment, and a second treatment was given with 5×106 cells/mouse, and then tumour burden was evaluated every 7 days.
Statistical analysis
All statistical analyses were performed using GraphPad Prism V.6.0 as described in the figure legends. Data are expressed as mean±SD. Student’s t-test was used for analyses between groups, and the difference was considered as ***p<0.001, **p<0.01 and *p<0.05.
Results
Generating BF minicircle vector
To develop a novel method for minicircle vector generation without using bacteria, we exploited simple molecular biology techniques of enzyme digestion and self-ligation reactions. Linear DNA fragments with compatible overhangs can be ligated either in transforming concatemers or in cis forming circles (figure 1, upper panel). When the concentration of DNA fragments is high, they tend to ligate in trans. While decreasing the concentration leads to higher cyclisation efficiency, the low concentration makes it unpractical to achieve high yield. To achieve higher cyclisation efficiency under higher DNA concentration, we developed a multiplexed strategy (figure 1). Ninety-six pairs of primers were designed to amplify the transgene, with each pair composed of 20 bp universal priming sequence, a 6 bp unique sequence and a BbsI enzyme recognition site at the 5’ end (online supplementary table S1). For PCR products generated using each pair of primers, BbsI digestion will generate compatible 4 bp overhangs at each end (figure 1, lower panel). During ligation, PCR products produced using different primer pairs will not ligate with each other, as their ends are not compatible. Therefore, each DNA fragment will more likely ligate in cis forming circles.
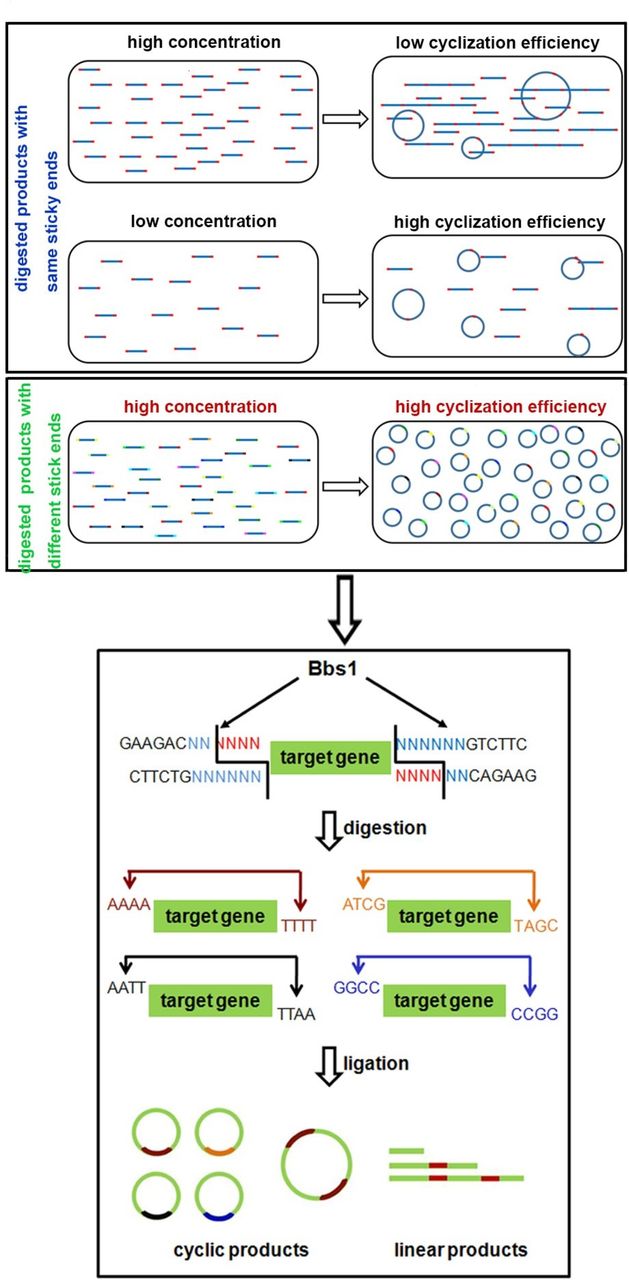
Schematic diagram of bacteria-free minicircle vector generation. The concentration of digested products affects the cyclisation efficiency of the minicircle molecule (upper panel) and the enzymatic digestion characterisation of BbsI (lower panel).
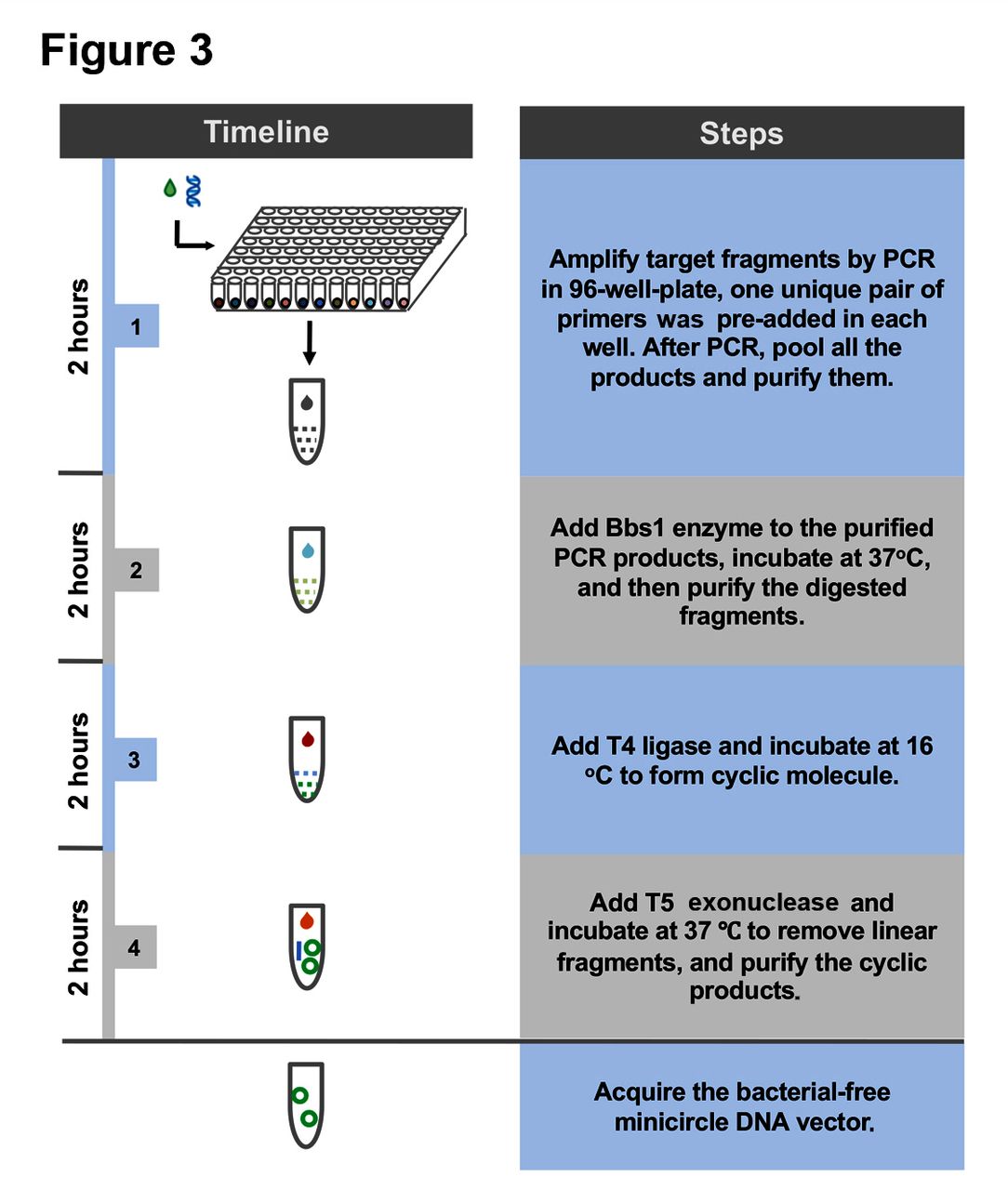
To validate this design, we prepared BF minicircle vectors expressing eGFP or CAR recognising CD19 (019-CAR). We amplified transgene using 96 pairs of primers independently, using multichannel pipette and 96-well PCR plate. After PCR amplification, all products were pooled, digested with BbsI and ligated. On gel electrophoresis, multiple bands were observed. After getting rid of all linear molecules using T5 exonuclease, the remaining band ran lower than the linear PCR product on the gel, indicating this band was the minicircle molecule (figure 2A). Indeed, after we treated this product with restriction enzymes that only cut once within the minicircle molecule, it was linearised into the correct size (online supplementary figure S1). We quantified the efficiency of minicircle molecule formation under different DNA concentrations. When the sample was amplified using only one pair of primers, the cyclisation efficiencies decreased rapidly with the increase of DNA concentration. In comparison, the cyclisation efficiencies of transgene amplified using 96 pairs of primers remained at a high level even when the DNA fragment concentration was increased (figure 2B and online supplementary figure S2). These results showed that we can generate minicircle vectors using a streamlined protocol within a few hours (figure 3).

Cyclisation of 019-CAR and eGFP transgene fragments. (A) Agarose gel electrophoresis of PCR fragments, and minicircle ligation products before and after T5 exonuclease treatment. (B) Cyclisation efficiencies of eGFP and 019-CAR transgene amplified with 1 pair of primers or 96 pairs of primers. Unpaired multiple two-tailed t-test. The asterisks in (B) indicate significant differences between 96 pairs of primers group with 1 pair of primers in each different DNA concentration: **p<0.01, ***p<0.001.

Flow chart of bacteria-free minicircle vector generation.
BF minicircle vectors enable robust transgene expression in cell line
We transfected BF minicircle vectors, linear PCR products and plasmids encoding eGFP to K562 cells by electroporation, and found that the cell viability in BF minicircle group was higher than that in plasmid group (online supplementary figure S3A). Compared with the rapid decline in PCR product group and plasmid group, both the percentage of eGFP-positive cells and the mean fluorescence intensity (MFI) of eGFP remained high in cells transfected with BF minicircle vector in the first 8 days, although they fell gradually thereafter (online supplementary figure S3B,C). From these data, we conclude that, compared with plasmid and linear DNA fragment, BF minicircle vector has the advantage of maintaining high cell viability and transgene expression level.
BF minicircle vector enables robust transgene expression in human CD34+ HSCs and ESCs
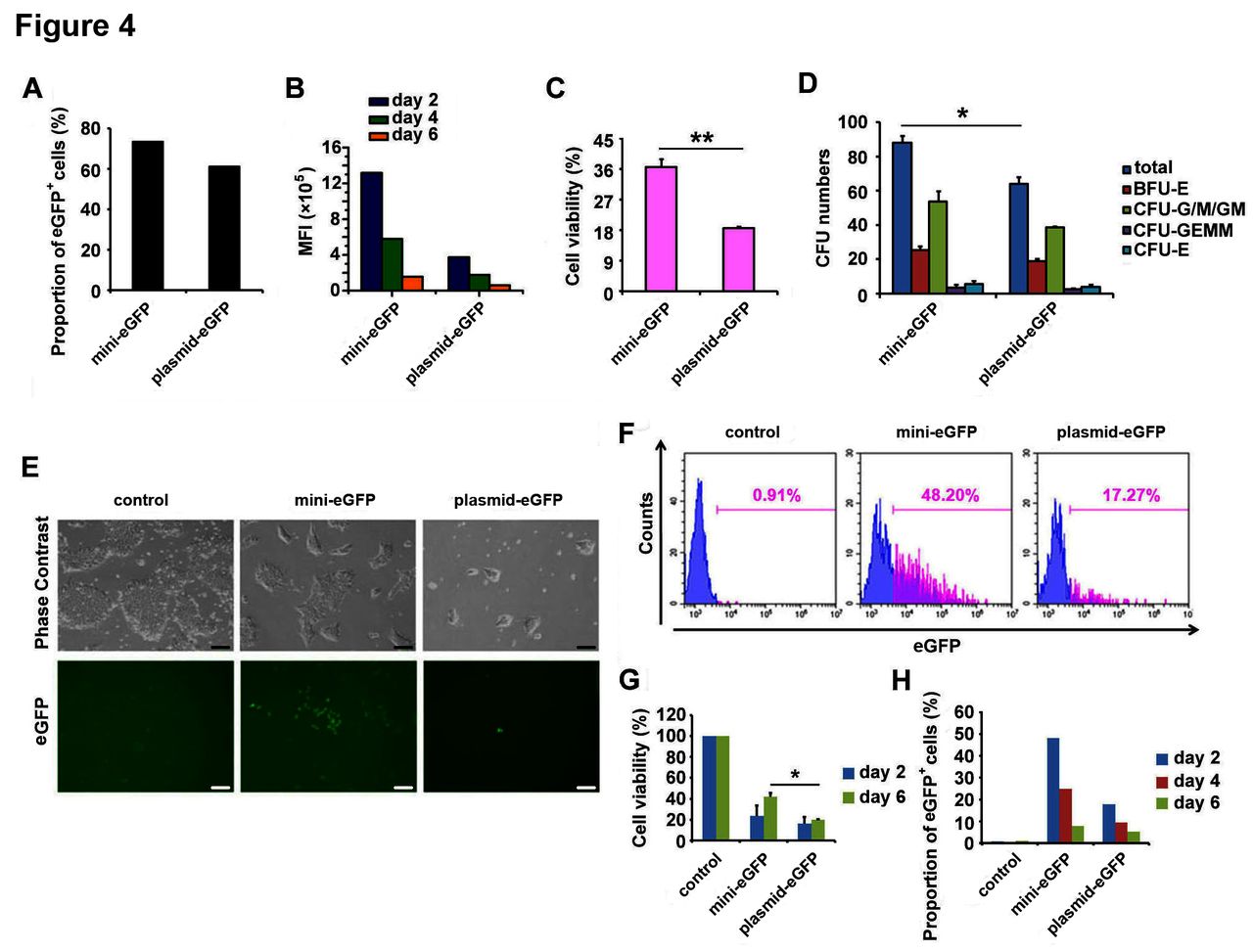
Next, we characterised the performance of BF minicircle vector in CD34+ HSCs isolated from umbilical blood. We found the MFI of eGFP in BF minicircle vector samples was much higher than in samples transfected with plasmid, while the eGFP-positive rates in both samples were similarly high (figure 4A,B). Meanwhile, the cell viability in BF minicircle vector electroporation group was significantly higher than the plasmid group (figure 4C). To characterise the differentiation potential of electroporated CD34+ HSCs, colony forming unit analysis was used. The electroporated cells formed colonies successfully, and erythroid, granulocyte, macrophage and megakaryocyte colonies were found in each group, suggesting that the stemness of HSCs was not affected (figure 4D).

BF minicircle vector electroporation in CD34+ HSCs and hESCs. (A) Proportion of eGFP-positive cells in CD34+ HSCs transfected with BF minicircle vector and plasmid encoding eGFP 48 hours after electroporation. (B) The MFI of eGFP-positive cells in days 2, 4 and 6 after mini-eGFP and plasmid-eGFP electroporation. (C) Cell viability of CD34+ HSCs transfected with BF minicircle vector and plasmid encoding eGFP 48 hours after electroporation. (D) Quantification of total CFU numbers and the numbers of BFU-E, CFU-G/M/GM, CFU-GEMM and CFU-E of CD34+ HSCs transfected with BF minicircle vector and plasmid encoding eGFP. (E) Typical images of H9 hESCs transfected with BF minicircle vector or plasmid encoding eGFP, 48 hours after electroporation. Scale bar=100 µm. (F) FACS analysis of eGFP-positive cells in H9 transfected with BF minicircle vector or plasmid encoding eGFP, 48 hours after electroporation. (G) Cell viability of H9 transfected with BF minicircle vector or plasmid encoding eGFP, 2 days and 6 days after electroporation. (H) Proportion of eGFP-positive cells in H9 transfected with BF minicircle vector or plasmid encoding eGFP, 2, 4 and 6 days after electroporation. (A–D) Data of CD34+ HSCs from one donor and more than three donor samples were detected. Unpaired multiple two-tailed t-test. The asterisks indicate significant differences between the mini-eGFP group and the plasmid-eGFP group: *p<0.05, **p<0.01. BF, bacteria-free; CFU, colony forming unit; eGFP, enhanced green fluorescent protein; FACS, fluorescence-activated cell sorting; HSC, haematopoietic stem cell; MFI, mean fluorescence intensity.
To evaluate the performance of BF minicircle vector in human ESCs, we electroporated BF minicircle vector and plasmid encoding eGFP into H9 cells.14 The eGFP expression and cell survival of BF minicircle vector-transfected cells were much better than that of plasmid (figure 4E–H). The H9 cells expressed OCT4 and NANOG uniformly, indicating that these cells were pluripotent (data not shown). All these data demonstrated that BF minicircle vector performed better than plasmid in stem cells, as evidenced by enhanced transgene expression level and cell viability.
Generating CAR-T cells using BF minicircle vectors
Next we tested whether we could generate transgene integration-free CAR-T cells using BF minicircle vector encoding 019-CAR and mediate antigen-specific cancer killing. We first compared the transgene expression levels of 019-CAR in primary CD3+ T cells transfected with BF minicircle vector and plasmid. T cells electroporated with plasmid survived poorly, while the cells in BF minicircle vector group showed a much higher viability and proliferating rate (figure 5A,B). BF minicircle vector enabled efficient CAR expression in primary T cells (figure 5C). These BF minicircle CAR-T cells (mini-019-CAR-T) secreted high levels of IL-2 and IFN-γ after incubation with CD19-positive tumour cells (figure 5D), and killed them at different effector to target ratios with an efficiency similar to CAR-T cells generated using lentivirus (lenti-019-CAR-T) (figure 5E).

Antitumour effects of mini-019-CAR-T cells in vitro. (A) T cells transfected with mini-019-CAR and plasmid-019-CAR. Photos were taken 48 hours after electroporation. Scale bar=100 µm. (B) Cell viability of T cells transfected with BF minicircle vector and plasmid on days 2, 4 and 6. (C) Expression level of 019-CAR in T cells transfected with BF minicircle vector, analysed by FACS analysis. T cells were isolated from umbilical blood from two different donors. (D) Concentrations of IFN-γ and IL-2 released by mini-019-CAR-T cells 24 hours after incubation with tumour cells. T cells were isolated from umbilical blood from two different donors. (E) Tumour cell lysis ability of 019-CAR-T cells prepared with BF minicircle vector (mini-019-CAR-T) or lentiviral vector (lenti-019-CAR-T) in different effector to target (E:T) ratios. T cells were isolated from umbilical blood from two different donors. T, T cells; K, K562 cell line; K19, K562 cell line overexpressing CD19. BF, bacteria-free; CART-T, chimeric antigen receptor T; GFP, green fluorescent protein; IFN-γ, interferon-γ; IL-2, interleukin-2.
As a control, we also generated 019-CAR minicircle vector (Kit-minicircle-019-CAR) using a commercially available minicircle vector producing kit (System Bioscience, USA), which is based on engineered bacteria strain and donor vector.9 Although we followed the manufacturer’s instruction rigorously and tried multiple times, the Kit-minicircle vector product was often contaminated with plasmid and genomic DNA to a certain extent (online supplementary figure S4A). After electroporation, the T cell survival rate in Kit-minicircle-019-CAR group was much lower than that in the BF mini-019-CAR group (online supplementary figure S4B), which might be due to the low purity of Kit-minicircle-019-CAR. We could not acquire enough Kit-minicircle-019-CAR-T cells for further function analysis.
BF minicircle CAR-T cells eradicated tumour efficiently in murine xenograft model
Next we compared the tumour alleviating capability of BF mini-019-CAR-T and lenti-019-CAR-T cells in a murine xenograft model. NPG mice were inoculated with Raji-luci cells intraperitoneally. Three days later, these mice were divided into four groups with similar tumour burden. T cells and CAR-T cells engineered with BF minicircle vector or lentiviral vector were administrated intraperitoneally on day 3 and day 9 (figure 6A). Tumour burden was monitored weekly by in vivo luciferase imaging. We found the tumour burden decreased significantly in mini-019-CAR-T-group and lenti-019-CAR-T-group, compared with that in the T cell group and PBS group (figure 6B,C). Impressively, the efficacy of mini-019-CAR-T is similar to lenti-019-CAR-T, indicating that CAR-T cells engineered with BF minicircle vector have robust tumour alleviating ability.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Antitumour effects of mini-019-CAR-T cells in vivo. (A) Experimental design of in vivo function test of CAR-T cells engineered with BF minicircle vector and lentiviral vectors. (B) Bioluminescent imaging result of NPG mice bearing Raji-luciferase tumour treated with T cells, lenti-019-CAR-T cells, mini-019-CAR-T cells or the same volume of PBS on days 3, 16 and 30 (n=3). (C) Quantification of luminescence at different time points. BF, bacteria-free; CAR-T, chimeric antigen receptor T; NPG, NOD-Prkdcscid Il2rgnull.
Discussion
Minicircle DNA molecule without any bacterial components is a valuable tool for basic research and gene therapy. Previous methods generating minicircle vector are time-consuming and require specific producer plasmids and bacteria strains. When we tried to produce minicircle vector using a commercial kit based on engineered bacterial strain, we could not produce minicircle vectors with high purity, and it took a few days to finish the procedure. Although this result could be due to our lack of experience with this kit, it showed the efforts required to use this system.
The method we established in this study allows for fast generation of minicircle vectors without using any bacteria culture, therefore eliminating any potential contamination from bacteria. The procedure to prepare BF minicircle can be summarised into five simple steps: (1) adding universal priming sequence to the transgene fragment by PCR; (2) PCR amplification of targeted gene fragment using 96 pairs of primers; (3) digestion of pooled PCR products by BbsI; (4) ligation of digested products by T4 ligase; and (5) removing linear products by T5 exonuclease (figure 3). With this method, we could acquire BF minicircle within 10 hours.
Although minicircle vector has been used to deliver Sleeping Beauty system to generate CAR-T cells,3 our study is the first to show that integration-free CAR-T cells generated using BF minicircle vector eradicated cancer cells efficiently both in vitro and in vivo. Adoptive transfer of engineered T cells has shown remarkable responses in patients with diseases such as cancer and viral infections.15 Transgene encoding T cells receptor (TCR) or CAR targeting tumour antigens are usually cloned in viral vectors. On viral transduction, transgenes expressing TCR or CAR are integrated into T cell genome. While transgene integration allows for constitutive expression, it also poses risk of insertional mutagenesis. In addition, the costly clinical-grade virus production is one of the major reasons for the high cost of clinical application. The recently FDA-approved CTL019 therapy (‘Kymriah’) has a price tag of $475 000. Our data suggest that transgene integration-free mini-019-CAR-T cells could be an attractive option for clinical applications. Although the length of transgene expression of BF minicircle vector is apparently shorter than lentivirus integrated in the genome, multiple rounds of infusion should be able to eliminate cancer cells efficiently, especially when a humanised CAR is used. On the other hand, a relatively shorter CAR-T persistence provides an opportunity to avoid serious side effects such as cytokine storm or on-target off-tumour toxicity.16
Using current protocol, the efficiency of mini-eGFP generation was about 30%–40% and mini-019-CAR was 20%–30%. Further optimisations, such as simplifying the preparation procedure, reducing product loss in each step and increasing cyclisation efficiencies of the DNA fragments, will be needed to achieve better yield of BF minicircle vector. Another caveat of our method is that the minicircle vector we generated is a mixture of 96 different molecules, each containing a unique 4 bp sequence at the ligation junction. Considering these 4 bp sequences are not being expressed and unlikely to affect transgene expression, this should not lead to any functional variations.
In conclusion, we established a simple and fast method to prepare minicircle vectors without using bacteria and generated functional mini-019-CAR-T cells. Compared with retrovirus or lentivirus, BF minicircle vector has the potential to improve safety and lower the production costs of CAR-T therapy and gene therapy.
Acknowledgments
We would like to thank Junning Wei and Yi Yang (Beijing Cord Blood Bank) for their help in preparing the cord blood samples. We would also like to thank Lei Shi (Beijing Cord Blood Bank) for her help with CFU analysis.
References
Footnotes
CC and NT contributed equally.
Contributors CC, NT and HW designed the experiments. CC, NT, JL, SC, TZ and XW performed the experiments. CC, NT and HW wrote the manuscript. All authors discussed and interpreted the results.
Funding This study was funded by the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no: XDA01010409, XDA16010205), State 863 project (grant no: 2015AA020307), National Natural Science Foundation of China (10.13039/501100001809) (grant nos: 31471215 and 81773269), and the National Key Research and Development Program of China (no 2016YFA0101402). HW is supported by the ’Young Thousand Talent Project'.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.