Article Text

Abstract
Background Several factors have been reported that influence the probability of a germline CDKN2A mutation in a melanoma family. Our goal was to create a scoring system to estimate this probability, based on a set of clinical features present in the patient and his or her family.
Methods Five clinical features and their association with CDKN2A mutations were investigated in a training cohort of 1227 Dutch melanoma families (13.7% with CDKN2A mutation) using multivariate logistic regression. Predefined features included number of family members with melanoma and with multiple primary melanomas, median age at diagnosis and presence of pancreatic cancer or upper airway cancer in a family member. Based on these five features, a scoring system (CDKN2A Mutation(CM)-Score) was developed and subsequently validated in a combined Swedish and Dutch familial melanoma cohort (n=421 families; 9.0% with CDKN2A mutation).
Results All five features were significantly associated (p<0.05) with a CDKN2A mutation. At a CM-Score of 16 out of 49 possible points, the threshold of 10% mutation probability is approximated (9.9%; 95% CI 9.8 to 10.1). This probability further increased to >90% for families with ≥36 points. A CM-Score under 16 points was associated with a low mutation probability (≤4%). CM-Score performed well in both the training cohort (area under the curve (AUC) 0.89; 95% CI 0.86 to 0.92) and the external validation cohort (AUC 0.94; 95% CI 0.90 to 0.98).
Conclusion We developed a practical scoring system to predict CDKN2A mutation status among melanoma-prone families. We suggest that CDKN2A analysis should be recommended to families with a CM-Score of ≥16 points.
- cancer: dermatological
- cancer: head and neck
- clinical genetics
- genetic epidemiology
- genetic screening/counselling
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- cancer: dermatological
- cancer: head and neck
- clinical genetics
- genetic epidemiology
- genetic screening/counselling
Introduction
Since its identification in 1994,1 the CDKN2A gene (MIM 600160) has remained the major high-risk susceptibility gene for cutaneous melanoma. Germline mutations are present in approximately 10%–40% of familial cases.2 Carriers of a germline mutation in the CDKN2A gene have an increased risk for developing melanoma, with a penetrance of up to 70% at 80 years of age, and 40% of carriers develop multiple primary cutaneous melanomas.3 Furthermore, mutation carriers have an increased risk for other types of malignancies, the most important of which is pancreatic cancer (PC).4 Due to the high risk of melanomas and other types of cancer and the advantages of regular surveillance in improving prognosis and survival,5 6 it is important to identify families that carry a CDKN2A germline mutation. However, the probability of a CDKN2A mutation strongly depends both on the clinical characteristics of a family and personal (dermatological) and environmental factors such as skin type and the amount of sun exposure. Thus, CDKN2A mutation analysis might not be indicated in some lower-risk melanoma families.
The Netherlands and Sweden both have a high incidence of melanoma (age-standardised rate 19.4 and 18.0 per 100 000, respectively)7 and specific founder mutations in the CDKN2A gene are the predominant cause of familial melanoma in these countries. In the Netherlands, the 19-base pair deletion termed p16-Leiden (c.225_243del, p.Ala76Cysfs*64; RefSeq NM_000077.4) confers an increased risk for melanoma and for tumours of the pancreas and upper airway tract (larynx, pharynx, oral cavity) and to a lesser extent tumours of the lungs and digestive tract.8–10 Carriers of the Swedish founder mutation (c.335_337dup, p.Arg112dup; RefSeq NM_000077.4) also show an increased risk for these tumours.11 12 Although it is recognised that the risk-spectrum for non-melanoma cancers differs among carriers of different mutations in the CDKN2A gene, pancreatic and upper airway tract cancers have repeatedly been reported in a variety of carrier populations.4 13–17
Over the past decade, research groups from Europe, USA and Australia have attempted to identify clinical features that are associated with germline CDKN2A mutations in melanoma families.18–24 Studied features included (1) number of patients with melanoma in a family, (2) number of patients with multiple primary melanomas (MPMs) in a family, (3) median age at diagnosis of melanoma and (4) presence of PC in a family. The most significant associations reported in these studies were the presence of more than two melanoma cases in a family, an early age of onset and having at least one family member with MPMs and/or PC. Based on a literature review from 2009, it was suggested that patients with melanoma from areas with a moderate to high incidence of melanoma are candidates for genetic testing of CDKN2A if they have at least three primary melanomas or when there are at least two additional diagnoses of melanoma and/or PC among close (first-degree or second-degree) family members (‘rule of threes’).25 The authors argued that these families have an estimated 10% or greater probability of carrying a germline CDKN2A mutation, which is a commonly used threshold in clinical practice for gene sequencing in hereditary cancer.26–28
The goal of this study was to create a scoring system for clinicians to estimate the probability of a germline CDKN2A mutation based on a set of clinical features present in the patient and his or her family. Using a training cohort of Dutch melanoma families, we therefore analysed the association of four previously reported clinical features that are associated with a CDKN2A mutation and investigated the association with upper airway cancer (UAC) as an additional feature. A combined cohort of Swedish and Dutch melanoma families was used for external validation of the scoring system.
Patients and methods
Training cohort
The training cohort included all index patients with cutaneous melanoma and their families in the Netherlands referred for CDKN2A mutation analysis between 1998 and 2015. According to current Dutch referral guidelines, CDKN2A mutation analysis is indicated if one of the following criteria is met: a family with (1) two first-degree relatives with melanoma, (2) two first-degree or second-degree relatives with melanoma and one first-degree or second-degree relative with PC, (3) three or more primary melanomas in one individual, (4) an individual with juvenile melanoma (<18 years) or (5) an individual with a history of both melanoma and PC. At the Department of Clinical Genetics at Leiden University Medical Centre, the Laboratory for Diagnostic Genome Analysis (LDGA) has been the primary sequencing facility for CDKN2A in the Netherlands since 1998 and receives diagnostic requests from across the Netherlands. Essential pedigree information was gathered for the families and added to the Leiden Familial Melanoma Database. These data included the number of first-degree and second-degree family members (of each other) with cutaneous melanoma (invasive or in situ), whether these patients had a single melanoma or MPMs, the age of each patient with melanoma at first diagnosis and the number of family members with PC and UAC, that is, cancer of larynx, pharynx or oral cavity. We restricted our analysis of these latter tumours to the first-degree and second-degree relatives of the index patient and the first-degree relatives of patients with melanoma. We relied on the referring clinical geneticists for complete pedigree information and, if necessary, histological confirmation of cancer diagnoses (melanoma and others). We included all information on cancer diagnoses and also those unconfirmed by the clinical geneticist, since index patient reports of melanomas in family members have a high known level of accuracy (true positive predictive value 77%–87%).29 We imputed the age of melanoma diagnosis for family members where the age at diagnosis was not reported in the pedigree (n=320 individuals from 212 families (61 with CDKN2A mutation)). Imputation was based on median age at diagnosis in CDKN2A mutation families (40 years) and in sporadic (non-CDKN2A) patients (55 years), as reported by van der Rhee et al.30 When the patient was younger than this age or was deceased prior to this age at time of CDKN2A analysis in the family, that specific age was used for imputation. Families without a CDKN2A mutation were excluded from the study if CDKN2A analysis was only performed in a non-affected family member (n=84). Families in which CDKN2A sequencing was unsuccessful were also excluded (n=4). The Leiden University Medical Centre Ethics Committee issued a declaration of no objection (#C14.064) regarding the creation of the Leiden Familial Melanoma Database.
Validation cohort
The greater portion of the validation cohort in this study consisted of members of melanoma-prone families from Sweden.31 Families were identified by questioning patients with newly diagnosed melanoma about their familial melanoma history. Melanoma families were defined as kindreds with at least two relatives (first-degree, second-degree or third-degree) with histologically or clinically verified melanoma. Since 1995, germline CDKN2A mutation analysis is offered to members of these families after informed consent is obtained. The study was approved by Research Ethical Review Boards at Lund University and Karolinska Institute in Stockholm, the sites where the genetic tests were performed. In Stockholm, patients with MPMs (regardless of family history) are also invited to undergo germline CDKN2A mutation analysis. In 2012, a study was performed to broaden understanding of the identified familial melanoma kindreds and of patients with MPMs through linkage to Swedish national registries.11 12 32 33 Further linkage to the Swedish Cancer Registry (established in 1958 with register completeness estimated to be 96%34) provided data on all registered cancers in the CDKN2A genotyped individuals and their first-degree and second-degree relatives.
Additional Dutch melanoma families were recruited at the Department of Dermatology, Leiden University Medical Centre, according to the inclusion criteria of the GenoMEL study (http://www.genomel.org/). After providing written informed consent, patients with melanoma were asked about their familial melanoma history. A melanoma family was defined by the presence of three or more cases with histologically confirmed melanoma or two cases with histologically confirmed melanoma in first-degree relatives.
DNA analysis
In the Dutch cohorts (both training and validation), DNA was extracted from whole blood samples of index patients and was used for sequencing of all coding exons of CDKN2A (1α, 1β, 2 and 3), including exon/intron boundaries. To detect larger deletions or duplications, multiplex ligation-dependent probe amplification was performed. In the early years of CDKN2A diagnostics, analysis was limited to a mutation-specific PCR for the detection of the p16-Leiden mutation. However, only a very small subset of CDKN2A wild type families in the training cohort was analysed in this manner (n=32). In an additional 89 families from the training cohort, exon 1β was not sequenced. For the Swedish cohort, procedures used for PCR of all CDKN2A exons and direct sequencing of PCR products have been described previously.11 Presence of a CDKN2A mutation was defined as having either a pathogenic or likely pathogenic variant in the CDKN2A gene (class 4 or 5 variant)35 or an unclassified variant (class 3) shown to be located on a pathogenic CDKN2A haplotype. Classification of these variants was based on (previously reported) cosegregation with disease, strong evidence of impaired protein function and, in some families, shared pathogenic haplotypes.
Statistical analysis
Five clinical features were predefined and used for analysis: (1) the total number of first-degree and second-degree family members (including the index patient) with a diagnosis of melanoma, (2) the number of these family members with MPMs, (3) the median age at diagnosis of (first) melanoma in the family and the presence of (4) PC and (5) UAC in a family. Median age at diagnosis was divided into three age groups (<30 years, 30–50 years and ≥50 years). A univariate analysis was performed to independently evaluate these features and a multivariate logistic regression model was used to assess the association between all five features and the presence of a germline CDKN2A mutation. The formula of the logistic regression model is P(robability)=eL/(1+eL) where L=constant + β1*C1 (number of family members with melanoma [1=0, 2=1, 3=2, ≥4=3]) + β2*C2 (number of family members with MPMs [0=0, 1=1,≥2=2]) + β3*C3 (median age at primary diagnosis [≥50=0,<50=1]) + β4*C4 (presence of PC [No=0, Yes=1]) + β5*C5 (presence of UAC [No=0, Yes=1]) and where β is the feature-specific β-coefficient. All statistical analyses were carried out in SPSS V.23.0.
Development of a scoring system: CM-Score
The β-coefficients derived from the multivariate analysis were converted to points for each feature using the formula Points=(Cx*βC)/B (as described by Sullivan et al,36 where Cx is the feature-specific numeral from the logistic regression formula, βC is the β-coefficient and B is the fixed multiplier or constant (defined 0.22). The total number of points was calculated for each family in the training cohort. Since there were often considerable differences in the number of families with successive point totals (for instance, there were 6 families with 21 points (33% mutation) and 37 families with 22 points (16% mutation)), the cohort was subsequently split into eight point-groups. This grouping would ensure a more accurate calculation of the observed mutation frequencies per group with narrower CIs. For each of these groups, the observed mutation frequencies, the mean of the predicted probabilities and their 95% CIs were calculated. The scoring system, CM-( CDKN2A Mutation) Score, was subsequently applied to the validation cohort, with the families split into the same point-groups as in the training cohort. The observed mutation frequencies and their 95% CIs were again calculated for each group. The performance of the scoring system was assessed for both the training cohort and the validation cohort with the Hosmer-Lemeshow goodness of fit test (calibration) and receiver operator characteristic (ROC) curve analysis with calculation of the area under the curve (AUC) (discrimination). The slope of the calibration line was estimated with linear regression. The proposed cut-off value in CM-Score for performing CDKN2A analysis was determined as the score that corresponds to a predicted mutation probability of ~10%.26–28
Results
Training cohort
A total of 1227 families were included in the study, 168 of which had a (likely) pathogenic variant in the CDKN2A gene (13.7%). The p16-Leiden founder mutation was present in 77% of these families (n=130) (online supplementary table S1). Most of the families had two or more members with melanoma (853 families; 70%) and included 503 two-case families, 233 three-case families and 117 families with four or more melanoma cases. In 654 (77%) of these multiple-case families, at least one additional clinical feature was present (ie, median age <50 years or presence of MPMs, PC or UAC in the family, see online supplementary table S2). In the 374 single-case families, 207 families (55%) had at least two other clinical features and 150 families (40%) had one other clinical feature. The majority of melanomas in the training cohort were confirmed by histology reports (76%). PC and UAC diagnoses were less frequently confirmed by the referring clinical geneticist (both 43%).
Supplementary file 1
Univariate and multivariate analysis
Having at least three family members with melanoma was significantly associated with the presence of a CDKN2A mutation in the univariate analysis (table 1). A median age of under 50 years and one or more cases with MPMs in a family were also significantly associated with a CDKN2A mutation. Age under 30 years at time of diagnosis did not result in a higher OR than age 30–50 years (OR 5.1 (95% CI 2.5 to 10.4) versus OR 7.1 (95% CI 4.1 to 12.3), respectively). A significantly increased risk for a CDKN2A mutation was seen in families in which PC and UAC co-occurred with melanoma; a mutation was present in 33% of the families with one or more patients with PC and 46% of the families with one or more patients with UAC.
Univariate analysis showing the independent association between each clinical feature and a germline CDKN2A mutation
In a multivariate logistic regression model, the five features investigated in the univariate model remained significantly associated with a mutation (table 2). Since in the univariate analysis, age under 30 years was not a stronger predictor than age 30–50 years, these age groups were combined into one group (age <50 years) for the multivariate analysis. The highest ORs were found for median age under 50 years (OR 8.5 (95% CI 4.5 to 16.0)) and for presence of PC or UAC in a family (OR 7.5 (95% CI 4.8 to 11.7) and OR 6.0 (95% CI 3.4 to 10.5), respectively), but these features had only two possible outcomes (<50 or ≥50 years, Yes or No), whereas the other melanoma-specific features had three or four possible outcomes and increasing ORs for each step.
Multivariate logistic regression model showing the association between all five clinical features combined and a germline CDKN2A mutation
CM-Score
The points assigned to each clinical feature are shown in table 3. The predicted mutation probabilities and observed mutation frequencies per point-group are shown in table 4. Below a total of 16 of 49 possible points, the predicted mutation probability is low (≤4.0%). Between 16 and 19 points, the predicted mutation probability is 9.9% and substantially increases in subsequent point-groups (20–23 points: 20.9%, 24–27 points: 34.7%, 28–31 points: 52.1%, 32–35 points: 71.4%,≥36 points: 90.7%).
Scoring system (CM-Score) based on the multivariate logistic regression model
Point totals from CM-Score with the corresponding mean predicted mutation probabilities and the observed mutation frequencies in the training and validation cohorts
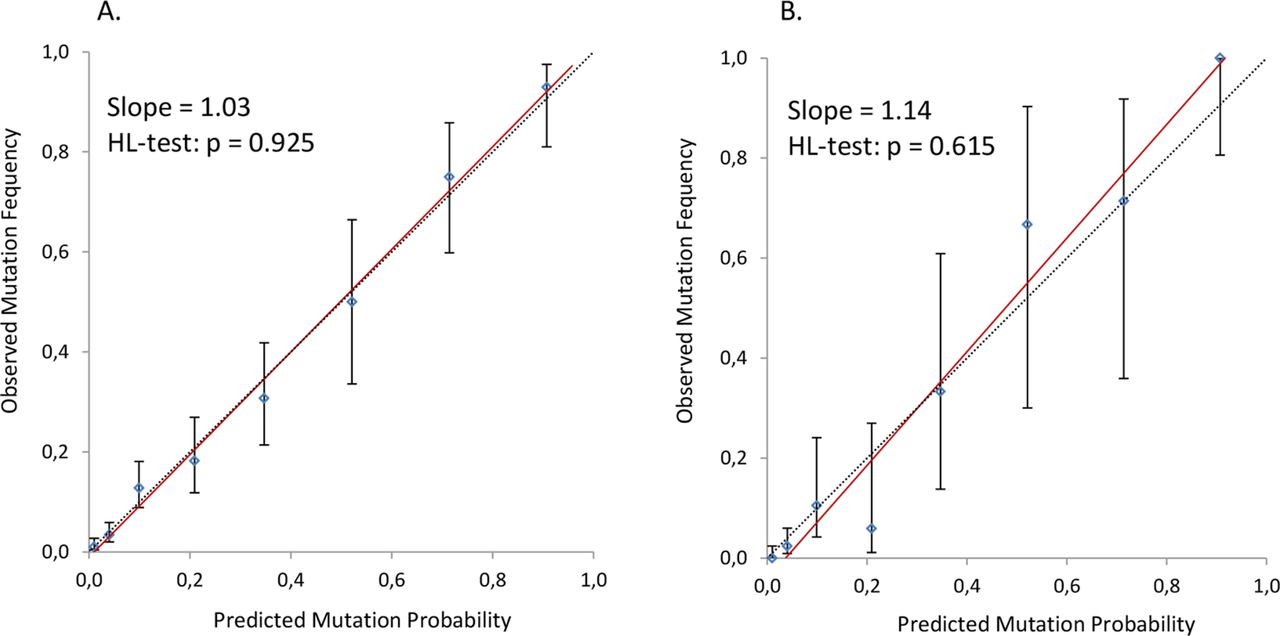
The concordance between observed and predicted mutation probabilities (calibration) is graphically displayed in figure 1A. The slope of the calibration line (1.03) indicates a good calibration, and the Hosmer-Lemeshow test (p=0.925) provided no evidence of a poor fit. Figure 2A shows the ROC curve analysis. The AUC is 0.89 (95% CI 0.86 to 0.92, p<0.001), which indicates that the model has a good ability to discriminate between families with and without a CDKN2A mutation. The threshold of 10% predicted probability is approximated at the cut-off value of 16 points in CM-Score, with a sensitivity of 90.5% (95% CI 84.7 to 94.2) and a specificity of 68.0% (95% CI 65.1 to 70.8). The majority of families (n=736; 60%) had a CM-Score of less than 16 points.

Calibration of CM-Score. (A) Training cohort. (B) Validation cohort. The calibration line (red) is a linear regression line that shows the relation between observed mutation frequency and predicted mutation probability in the training cohort (A) and the validation cohort (B). The dashed line is the reference line of perfect calibration. The 95% CIs of the observed mutation frequencies per point-group are displayed by the vertical lines. HL-test, Hosmer-Lemeshow test.

{kind=link}
{kind=link}
Discriminative ability of CM-Score. (A) Training cohort. (B) Validation cohort. ROC curve analysis of the training cohort (A) and the validation cohort (B). Point total was used as the test variable and mutation status was used as the state variable. Comparable results were obtained when the calculated predicted probability was used as test variable. AUC, area under the curve; ROC, receiver operator characteristic.
External validation of the scoring system
The validation cohort consisted of a total of 421 families (403 from Sweden; 18 from the Netherlands), of which 38 had a (likely) pathogenic variant in the CDKN2A gene (9.0%). Most of these families (n=30; 79%) carried the Swedish founder mutation p.Arg112dup and two Dutch families carried the p16-Leiden founder mutation (online supplementary table S3). The majority were multiple-case families (294 families; 70%) and included 232 two-case families, 37 three-case families and 25 families with four or more melanoma cases. All melanomas in the validation cohort were histologically confirmed. PC was present in 29 families (28 histologically confirmed; 72% CDKN2A mutation) and UAC in 24 families (23 histologically confirmed; 63% CDKN2A mutation).
The observed mutation frequencies per point-group in the validation cohort are shown in table 4. The performance of CM-Score in the validation cohort is displayed in figures 1B and 2B. The slope of the calibration line is 1.14 with a non-significant Hosmer-Lemeshow test (p=0.615). The AUC is 0.94 (95% CI 0.90 to 0.98, p<0.001), indicating good performance of CM-Score in the validation cohort. The sensitivity and specificity at the cut-off value of 16 points is 89.5% (95% CI 74.3 to 96.6) and 83.8% (95% CI 79.6 to 87.3), respectively. Similar to the training cohort, the majority of families in the validation cohort (n=325; 77%) had a CM-Score of less than 16 points.
Discussion
This study in a large Dutch training cohort of 1227 melanoma families confirmed the importance of four previously established clinical features that are associated with the presence of a germline CDKN2A mutation in a patient with melanoma. Furthermore, a fifth feature, the presence of UAC in the family, could be validated. Based on these clinical features and their ORs in our multivariate logistic regression model, we developed the CM-Score system to predict CDKN2A mutation probability, which performed very well in a combined Swedish and Dutch external validation cohort (AUC 0.94). At a cut-off value of 16 out of 49 points, the predicted probability approximates the commonly used 10% predicted probability threshold for germline gene sequencing in hereditary cancer, with a sensitivity of 89% and a specificity of 84% in the validation cohort. This cut-off value is also clinically relevant, since the majority of families in the training and validation cohorts scored less than 16 points (60% and 77%, respectively), a threshold below which the probability of a mutation decreases substantially (≤4%). Use of CM-Score could potentially spare many families (extensive) genetic testing, which may be particularly relevant in countries where resources for genetic testing are limited. Conversely, in families with a high CM-Score and therefore high mutation probability, genetic testing is even more urgent. A scoring system should, however, always only complement the clinical judgement of the clinical geneticist requesting DNA diagnostics (for instance, taking into account family size, age of family members, whether a patient has a certain combination of different malignancies and the availability of reliable medical information).
Risk models involving melanoma37 and CDKN2A mutation probability23 24 have been described previously. Niendorf et al incorporated the features (1) number of primary proband melanomas, (2) number of primary melanomas in the family and (3) age in a logistic regression model they named MELPREDICT.23 The AUC was 0.881 in the training set (n=116 families) and 0.803 in the external validation set (n=143 families). A computerised optimisation of this model, renamed MelaPRO, was published in 2010 and outperformed the former model with an AUC of 0.86 in a validation set of 167 families.24 MelaPRO includes the same clinical (familial) features as MELPREDICT, but also takes into account regional melanoma incidence rates and the geographical penetrance of CDKN2A. In contrast, while our CM-Score was trained and validated using families of Northern European descent, its strength lies in its simple, non-computerised scoring system that incorporates five features (including the presence of PC and UAC in a family) and despite this simplicity shows a superior performance in very large sets of melanoma-prone families.
The guidelines for CDKN2A mutation testing proposed by Leachman et al in 200925 were recently updated.38 In view of the recent reports of non-CDKN2A melanoma syndromes, such as those related to germline mutations in BAP1 39 (MIM 603089), POT1 40 (MIM 606478) and MITF 41 (MIM 156845), the authors propose tailored multigene panel testing in melanoma families instead of CDKN2A mutation testing alone. The 2009 criteria for genetic testing were converted into a points system, with points awarded for cancers that occur in so-called melanoma-dominant syndromes and melanoma-subordinate syndromes (where melanoma is not the predominant cancer type, such as in hereditary breast and ovarian cancer). Based on these points, the clinical geneticist can subsequently select the appropriate gene panel(s) to be tested in a family. In the selection and genetic assessment of melanoma families, this is a rather different approach to the one we propose in the current study. First, CM-Score is designed for families where melanoma is the predominant cancer type. Second, since CDKN2A is still by far the major susceptibility gene in familial melanoma, we based the selection of families for genetic assessment on the probability of specifically detecting a CDKN2A mutation in these families. Because other melanoma-dominant syndromes (such as those related to BAP1, POT1, CDK4 and MITF) are very rare compared to CDKN2A-related familial melanoma (each gene contributing <1%),42 we hypothesise that the calculated mutation probability from CM-Score largely reflects the joint probability of detecting a germline CDKN2A mutation or any other melanoma-dominant mutation. However, it should be noted that some tumours that are not part of CM-Score are highly specific to non-CDKN2A melanoma syndromes, especially BAP1-related tumours such as uveal melanoma and mesothelioma.43 44 BAP1 germline analysis should therefore be specifically offered when these tumours co-occur with cutaneous melanoma in a family, either together with CDKN2A or as part of a multigene panel test. It is not within the scope of this study to elaborate on the choice between multigene panel testing and CDKN2A mutation testing alone in melanoma-prone families. Although multigene panel testing increases the detection rate of cancer-predisposing germline mutations, there is also an elevated risk of identifying a variant of unknown significance in one of the genes and therefore increasing the uncertainty for a family regarding their genetic risk. The chance of this happening increases as more genes are included in a panel or when multiple panels are considered. Pros and cons of multigene panel testing should therefore always be carefully discussed with the patient.
Strengths of our study include relatively large and homogeneous cohorts and the broad analysis of five clinical features, including one more recently described feature (ie, UAC). However, because the scoring system is based on populations with a high melanoma incidence, it is possible that it will underestimate the probability of finding a CDKN2A mutation in lower melanoma incidence areas such as Southern (Mediterranean) Europe or overestimate the probability in extreme incidence areas such as Australia. Additional validation in other geographical areas would therefore be valuable. Another limitation of our study is information bias. In the training cohort, we had to rely on information supplied by the referring clinical geneticists and not all melanoma diagnoses were therefore histologically confirmed (76%). However, since the reporting of additional melanomas in family members by the index patient is known to be highly accurate, this factor is unlikely to have influenced the results.29 Unfortunately, only 43% of all pancreatic tumours and 43% of all upper airway tumours were confirmed. Nevertheless, all melanomas and other cancers in the validation cohort were verified since the majority of diagnoses were derived from the Swedish Cancer Registry.
In conclusion, we have developed and validated a non-computerised and clinically easy-to-use scoring system that shows high utility in predicting the probability of a germline CDKN2A mutation in melanoma-prone families from Northern Europe. The scoring system is based on clinical information on melanoma diagnoses in the patient’s family and additionally includes diagnoses of PC and UAC. As CM-Score was trained and validated in large sets of Northern European families, we suggest that the system should be further validated in other regions as well. In view of the 10% mutation probability threshold, we suggest that CDKN2A analysis should be recommended to families with a CM-Score of ≥16 points.
Acknowledgments
We are indebted to the participating families, whose generosity and cooperation have made this study possible. We acknowledge the contributions to this work made by Diana Lindén, Lena Westerberg, Anita Schmidt-Zander, Rainer Tuominen and Johan Hansson. We thank Medactie.com for help with editing of this paper.
References
Footnotes
Contributors Design and conception of study: TPP, HFAV, OMD, FJH. Data collection and assembly: TPP, HH, ML, NvdS, NAG, VH, HO. Data analysis and interpretation: TPP, HH, ML, RvD, HFAV, CJvA, OMD, FJH. Writing of manuscript: TPP, HH, ML, FJH. Critical review and revision of manuscript: all authors. Submission of manuscript: TPP.
Funding This work was supported by the Dutch Cancer Society (UL 2015-7511 and UL 2012-5489); The Swedish Cancer Society (CAN 2013/637, CAN 2014/851 and CAN 2015/283); Genomel (LSHC-CT-2006–018702); ERC advanced grant 2011 (291576); The Radiumhemmet Research Funds (144073) and Regional Funds and Hospital Funds in Lund and Stockholm.
Competing interests None declared.
Patient consent Not required.
Ethics approval Leiden University Medical Centre Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Collaborators Dutch Working Group for Clinical Oncogenetics (participating members): A Wagner, L van der Kolk, M Ausems, Th Van Os, E M Leter, L Spruijt, K van Engelen, L Berger.