Article Text

Abstract
Background Hereditary sensorineural hearing loss is a genetically heterogeneous disorder.
Objectives This study was designed to explore the genetic etiology of deafness in a large Chinese family with autosomal dominant, nonsyndromic, progressive sensorineural hearing loss (ADNSHL).
Methods Whole exome sequencing and linkage analysis were performed to identify pathogenic mutation. Inner ear expression of Ifnlr1 was investigated by immunostaining in mice. ifnlr1 Morpholino knockdown Zebrafish were constructed to explore the deafness mechanism.
Results We identified a cosegregating heterozygous missense mutation, c.296G>A (p.Arg99His) in the gene encoding interferon lambda receptor 1 (IFNLR1) – a protein that functions in the Jak/ STAT pathway– are associated with ADNSHL. Morpholino knockdown of ifnlr1 leads to a significant decrease in hair cells and non-inflation of the swim bladder in late-stage zebrafish, which can be reversed by injection with normal Zebrafish ifnlr1 mRNA. Knockdown of ifnlr1 in zebrafish causes significant upregulation of cytokine receptor family member b4 (interleukin-10r2), jak1, tyrosine kinase 2, stat3, and stat5b in the Jak1/STAT3 pathway at the mRNA level.
Conclusion IFNLR1 function is required in the auditory system and that IFNLR1 mutations are associated with ADNSHL. To the best of our knowledge, this is the first study implicating an interferon lambda receptor in auditory function.
- ADNSHL
- deafness gene
- IFNLR1
- JAK/STAT pathway
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Hereditary hearing loss is characterised by a high degree of genetic heterogeneity with mutations in several hundreds of genes encoding a variety of proteins.1 Individuals with autosomal-dominant non-syndromic hearing loss (ADNSHL), comprising ~20% of those with inherited hearing loss, typically display postlingual progressive hearing impairment that initially involves hearing loss of high frequencies. To date, 59 ADNSHL genetic loci have been mapped, and 36 causative genes have been identified (http://hereditaryhearingloss.org/). Based on the type of gene product, these genes can be categorised into several groups of encoded proteins such as those involved in the structure and function of hair cells, the auditory nerve and virtually every structural element of the inner ear. The pace of gene discovery has accelerated with the increased availability of high throughput sequencing techniques.
Interferon (IFN) lambda receptor 1 (IFNLR1, MIM 607404) belongs to the class II cytokine receptor family, which is responsible for recognition of cytokines and IFNs in the extracellular environment and initiation of intracellular signalling cascades that lead to an array of responses such as haematopoiesis, regulation of the immune system and cellular growth and development.2 3 The functions of IFNλ include antiviral activity,4 5 antiproliferative effect,6 antitumor activity7 and the regulation of major histocompatibility complex class I/II molecule expression8 and immune responses.9 10 Three research groups identified IFNLR1, so it has three alternative names: CRF2-12, IL28R and LICR.4 5 11 However, the function of IFNLR1 in the auditory system has not been described.
Here, we used a combination of genetic linkage analysis and whole-exome sequencing to identify a mutation in IFNLR1 as the cause of an autosomal-dominant hearing impairment. We further evaluated the expression of this gene in mice and verified that its disruption in zebrafish leads to auditory organ impairment and developmental defects.
Methods
Ethics statement
We obtained fully informed written consent from adult subjects and parents of minor subjects for participation in this study and publication of their clinical data.
Family ascertainment and clinical evaluation
Participating subjects were evaluated by medical history interviews, and a physical examination was performed on index patient from family JS4842. The degree of hearing loss was assessed using pure-tone audiometry, auditory brainstem response (ABR) and distortion product otoacoustic emission (DPOAE). CT scans of temporal bones were performed. Vestibular functions were evaluated using the tandem gait, Romberg test and caloric test. The index patient was further subjected to an ophthalmological examination with funduscopy and other tests including serum chemistry analysis, blood count, urinalysis and an ECG. Genomic DNA was extracted from peripheral blood using a blood DNA extraction kit according to the manufacturer’s instructions (TianGen, Beijing, China).
Whole-exome sequencing
Whole-exome capture and sequencing was performed (II:1, II:4, III:4, III:5, III:6, III:7, figure 1A). DNA was sheared, ligated to adaptors, extracted, amplified by ligation-mediated PCR and then hybridised to the Nimblegen SeqCap EZ Exome Kit V.3.0 (Roche, Basel, Switzerland) for enrichment. Each captured library was loaded onto the Illumina Hiseq2000 platform. After filtering out low-quality and duplicate reads, clean data were aligned to the human reference genome hg19 using the Burrows-Wheeler Aligner. After alignment, variants were called using four types of software (SOAPsnp, GATK, Samtools and Platypus), merged into variant call format files and annotated using a variant effect predictor, including those with minor allele frequencies (MAF), in public databases and the Beijing Genomics Institute (BGI) in-house databases. We used ExomeDepth software to detect the CNVs of this pedigree. By setting the parameters and following the instructions, we obtained rare CNVs from exome sequencing data. Under the assumption of autosomal-dominant pattern of inheritance, only variants that were heterozygous in the four affected siblings and absent in the two unaffected individuals were selected as candidates and verified using PCR-Sanger sequencing.

Identification of a mutation in IFNLR1 causing ADNSHL. (A) The pedigree of a three-generational family with segregating progressive ADNSHL. DNA samples were obtained from six unaffected and nine affected individuals (denoted by asterisks). Individuals who underwent whole-exome sequencing and linkage analysis are marked with W. Black symbols represent affected individuals. The haplotype created with SNP markers shows complete cosegregation of a locus at 1p34.1–1 p36.12 (red) in family JS4842. (B) Distribution of variants in the chromosome. Blue represents the distribution of somatic SNVs, orange indicates the distribution of somatic Indel mutations and grey represents the distribution of somatic CNVs. (C) Schematic diagram of IFNLR1 and protein alignment showing IFNLR1 Arg99His occurred at evolutionarily conserved amino acids (in red box) within the tissue_fac domain across nine species. (D) Electropherograms showing that the IFNLR1 c.296G>A mutation cosegregated with hearing loss. (E) Audiograms from selected individuals of family JS4842. Thresholds reveal the presence of moderate to severe sensorineural hearing loss, which occurred at a late onset in all affected individuals. For clarity, thresholds are shown for one ear from each individual. No differences in hearing were detected when the two ears were compared in an affected individual. Younger individuals had less severe hearing loss compared with the older affected individuals, suggesting that hearing loss in this family is progressive in nature. ADNSHL, autosomal-dominant, non-syndromic, progressive sensorineural hearing loss; IFNLR1, interferon lambda receptor 1; SNV, single nucleotide variant.
Linkage analysis and haplotype analysis
We performed a genome-wide linkage analysis of 15 family members (marked with asterisks in figure 1A) using the Human Omni ZhongHua-8 Bead chips (Illumina, San Diego, California, USA) containing 900 015 SNP markers. A total of 10 566 Tag SNPs distributed every 0.3 cM of genomic DNA were chosen to calculate the logarithm (based 10) of odds (LOD) scores using the Merlin (V.1.1.2) parametric linkage analysis package. The complete penetrance autosomal-dominant model was used with a rare disease frequency of 0.0001. Critical recombination events of the pedigree were also determined through haplotype construction using the Merlin (V.1.1.2) software.
Mutation analysis
Direct sequencing was used for mutation screening. Primers were designed to amplify all IFNLR1 exons and ~100 bp of adjacent intronic sequence flanking the exons (primer sequences available on request). A total of 500 negative control samples with normal hearing and 97 individuals from ADNSHL cohorts with sloping audiograms were sequenced for mutations in IFNLR1.
Model building and multiple sequence alignment
Three-dimensional modelling of p.Arg99His was performed using SWISS-MODEL, an automated homology modelling program (http://swissmodel.expasy.org/workspace/). Data obtained by the homology models were visualised using Swiss-PdbViewer 4.1. Multiple sequence alignment was performed according to a Homologene program.
Immunostaining
Slides spanning the entire inner ear from three different mice were stained, and the observed expression patterns were considered reliable if present in all three samples. For cryosections, the ears of C57 mice were fixed with 4% paraformaldehyde, decalcified, dehydrated with a graded series of ethanol; serial sections were cut at 8 µm thickness. An anti-Ifnlr1 antibody was diluted (1:200 rabbit polyclonal antibody; Bioss, Beijing, China), and Phalloidin-iFluor 555 (1:400, Abcam, Cambridge, UK) was added for actin staining. The nuclei were labelled with DAPI (4',6-diamidino-2-phenylindole, 1×, ZSJB-Bio, Beijing, China), and the primary antibody was omitted as a control. Sections were examined using confocal microscopy. For immunostaining of whole-mount organ of Corti, mouse cochlea ducts were dissected, fixed and incubated with anti-Ifnlr1 (1:200) and DAPI and analysed using a confocal microscope (Zeiss).
Zebrafish husbandry and iflnr1 Morpholino (MO) knockdown
AB line zebrafish were used and maintained at the PLA general hospital Zebrafish Breeding Centre. Tg (Brn3c:mGFP) S356T transgenic zebrafish expressing GFP in hair cells under control of the POU4F3 promoter that is targeted to the plasma membrane by a GFP-43 membrane targeting sequence (provided by Professor Hua-Wei Li, Fudan University) were used. Antisense MO (Gene Tools, Philomath, Oregon, USA) was microinjected into fertilised one-cell-stage embryos according to standard protocols.12 The sequence of the exon 4-intron 4 splice region of ifnlr1 MO (ifnlr1-e4i4-MO) was 5′-AGAGATGATACTAACCTGTGATCCG-3′, and the sequence for the standard control MO was 5′-CCTCTTACCTCAGTTACAATTTATA-3′ (Gene Tools). For the ifnlr1 gene knockdown experiment, 4 ng of control-MO or ifnlr1-e4i4-MO (E4I4-MO) were used per injection. Total RNA was extracted from 30 to 50 embryos per group in TriPure Isolation Reagent (Roche, Indianapolis, Indiana, USA) according to the manufacturer’s instructions. RNA was reverse transcribed using the PrimeScript RT reagent Kit with gDNA Eraser (Takara, Otsu, Japan). Primers spanning ifnlr1 exon 3 (forward primer: 5′-ACAGTGTCTGGGTGAAGTG-3′) and exon 5 (reverse primer: 5′- GAAGGAGACGTCCTGCTAAA-3′) were used for RT-PCR analysis to confirm the efficacy of the E4I4-MO. The ef1α primer sequences used as the internal control were 5′-GGAAATTCGAGACCAGCAAATAC-3′ (forward) and 5′-GATACCAGCCTCAAACTCACC-3′ (reverse).
Immunofluorescence staining and cell proliferation assays
Four days postfertilisation (dpf) MO and WT larvae were incubated in EM (Embryomedium) containing 15 mM bromodeoxyuridine (BrdU; Sigma-Aldrich) for 48 hours at 28.5°C. The larvae were then fixed in 4% PFA (Paraformaldehyde) for 4 hour at room temperature (RT) or overnight at 4°C and washed three times in PBST with 0.1% TritonX100 (PBST). All larvae were placed into 2 n HCl for 1 hour at RT. Larvae were washed again in PBST and blocked in 10% normal goat serum in PBST for 1 hour at RT. The larvae were incubated in the primary antibody, including the Chicken anti-GFP (1:200, Abcam, ab13970), rabbit monoclonal anti-sox2 (1:200, Abcam, ab137385), mouse anti-BrdU IgG (1:200, Santa Cruz Biotechnology, Dallas, Texas, USA) overnight at 4°C, and the secondary antibody including Alexa Fluor 488 anti-Chicken (1:200, Life technologies, A11008), Cy3 anti-Mouse (1:200, Jackson ImmunoResearch Laboratories, West Grove, PA, USA), Alexa Fluor 647 anti-Rabbit (1:200, Abcam, ab150075) was used at 4°C overnight. DAPI were used at R for 20 min. Cells were counted under a confocal microscope.
Acridine orange staining for apoptosis
Control MO-injected embryos and embryos injected with 4 ng ifnlr1-e4i4-MO were immersed in 5 µg/mL acridinium chloride hemi-(zinc chloride) (AO; Sigma-Aldrich, St. Louis, Missouri, USA) in fish water for 60 min at 3 dpf. Zebrafish were then oriented on their lateral side and mounted with methylcellulose in a depression slide for observation by fluorescence microscopy.
In vivo macrophage migration assays in zebrafish
To evaluate macrophage migration in zebrafish, fertilised one-cell (zlyz:EGFP) transgenic (TG) line embryos were injected with 4 ng ifnlr1-e4i4-MO or control-MO. At 6 dpf, the number of macrophages recruited to the body trunk was counted.
DASPEI staining of lateral line hair cells
After microinjection, larvae (6 and 7 dpf) were immersed in 1 mM 2-(4-(dimethylamino)styryl)-N-ethylpyridinium iodide (DASPEI; Sigma-Aldrich, Oakville, Ontario, Canada) in fish water for 1 hour. larvae were then oriented on their lateral side (anterior, left; posterior, right; dorsal, top) and mounted with methylcellulose in a depression slide for observation by fluorescence microscopy.
Quantitative real-time PCR
Total RNA was extracted from 80 to 100 embryos per group in Trizol (Roche, Indiana, USA) according to the manufacturer’s instructions. RNA was reverse transcribed using the PrimeScript RT reagent Kit with gDNA Eraser (Takara, Otsu, Japan). Quantification of gene expression was performed in triplicate using Bio-Rad iQ SYBR Green Supermix (Bio-Rad, Hercules, California, USA) with detection on the Realplex system (Eppendorf). Relative gene expression quantification was based on the comparative threshold cycle method (2 − ΔΔCt) using ef1α as an endogenous control gene. Primer sequences are available on request.
pcDNA3-ifnlr1 recombinant vector construction
The ifnlr1 cDNA was reverse transcribed from mRNA from zebrafish embryos. The complete ifnlr1 cDNA was subcloned into pcDNA3 to construct the recombinant vector pcDNA3-ifnlr1.
Image acquisition and statistical analysis
Embryos and larvae were analysed using a Nikon SMZ 1500 fluorescence microscope and subsequently photographed with a digital camera.
All data are presented as mean±SEM. Statistical analysis and graphical representation of the data were performed using GraphPad Prism 5.0 (GraphPad Software, San Diego, California, USA). Statistical significance was calculated using Student’s t-test, one-way analysis of variance (ANOVA) or χ2 test, as appropriate. Statistical significance is indicated as *P<0.05 and ***P<0.0001.
Results
Clinical findings
We examined a three-generation Chinese family (No. JS4842; figure 1A) with segregating autosomal-dominant, non-syndromic, bilateral, symmetric, sensorineural hearing loss that begins in the second decade of life, generally accompanied by high-frequency tinnitus. The youngest affected family member, III:6, was diagnosed at 21 years of age with hearing loss involving mid-high frequency, while his previous audiogram was normal at 19 years of age; therefore, the age at onset of hearing loss could be determined. The affected individuals reported a progressive nature of hearing loss, which was verified with audiograms. While affected family members initially had normal low-frequency hearing, their hearing loss became progressive and ultimately deteriorated at all frequencies by the fourth or fifth decade (figure 1E). There were no indications of a relationship between hearing loss and infections or noise. Result of ABR and DPOAE excludes auditory neuropathy spectrum disorders. All affected individuals did not complain of imbalance, and no abnormalities were found in a detailed vestibular analysis. The affected individuals had no obvious delay in gross motor development and no indications of visual problems or morphological features to suggest a syndromic form of hearing loss. High-resolution CTs of the temporal bone in affected individuals were normal, excluding inner-ear malformations.
Linkage analysis, whole-exome sequencing and in silico analysis
DNA samples were obtained from six unaffected and nine affected individuals of family JS4842. One affected individual (III:5) was prescreened and found to be negative for mutations in GJB2, SLC26A4 and mtDNA12SrRNA via direct sequencing.
Linkage and haplotype analysis mapped the pathogenic gene in this family to one loci, a shared region of 21.45 Mb on chromosomal region 1p34.1–1 p36.12 (IFNLR1 1p36.11), located between rs2286243 and kgp9850329 (figure 1A); this region corresponded to the 21.45 Mb genomic region chr1:22,986,610 to chr1:44,441,533 (hg37) and included 289 genes. The LOD scores ranged from 3.0237 to 3.3109. This linkage interval overlapped with that reported for the dominant non-syndromic deafness locus, DFNA2.13–15
To identify the gene responsible for hearing loss in the family, we performed whole exome sequencing in four affected and two unaffected subjects. We obtained 49 229 SNPs and an average of 5456 InDels (figure 1B) located on the exon target region, and we filtered out the variants with MAF >0.005 from public databases. These variants were shared by the four cases and were absent from two controls. In the 21.45 MB region, After the PCR-Sanger sequencing, we found two variants, c.296G>A (p.Arg99His) (figure 1D) and CDC20 c.1436G>A (p.Arg479Gln), that cosegregated in the phenotype and genotype in this pedigree. All nine affected individuals were heterozygous for these mutations. We further expanded the BGI in-house database and checked the frequency of the two variants. CDC20 c.1436G>A (p.Arg479Gln) was carried by eight individuals who were diagnosed with retinitis pigmentosa but reported no hearing loss. IFNLR1 c.296G>A was absent from 2896 in-house controls in Chinese population. We detected an average of 346 CNVs per sample, but none met the genetic model of this pedigree. PCR-Sanger sequencing of the variant in 500 normal-hearing controls having the same genetic background revealed no carriers of IFNLR1 c.296G>A (p.Arg99His) and this residue is conserved in all vertebrates, from zebrafish to mammals (figure 1C). The mutation is likely to affect protein function, as predicted by several models including SIFT (deleterious), PolyPhen-2 (probably_damaging), PhastCons (disease causing), Mutation Assessor (medium impact), FATHMM (tolerated) and GERP++ (damaging).
The p.Arg99His molecular model covered the target sequence of IFNLR1 (residues 26–221) and was constructed based on the crystal structure of IFNLR1 (PDB ID: 5t5w.1.B). The sequence identity between the target and template was 100%. Using Swiss-Pdb Viewer 4.1, the mutation was predicted to perturb the amino acid side chain due to the substitution of arginine to histidine at position 99 (online supplementary figure 1).
Supplementary file 1
To determine the possible involvement of IFNLR1 mutations in the Chinese hearing loss population, all seven exons of IFNLR1 were screened via Sanger sequencing in 96 index patients from ADNSHL families of Chinese Han ethnicity; no other mutations were identified.
Localisation of Ifnlr1 in mouse inner ear
Figure 2A presents a schematic overview of the Ifnlr1 expression pattern in the inner ears of mice at P30. In the cochlea, Ifnlr1 was expressed in the organ of Corti, Reissner’s membrane, basilar membrane, the spiral ligament, inner sulcus cells, inner pillar cells and outer pillar cells (figure 2B). The expression of Ifnlr1 was enriched in the stereocilia of both inner hair cells (IHCs) and outer hair cells (OHCs), displaying partial colocalisation with phalloidin (figure 2G). An antibody directed against the heavy chain of neurofilament (NF200), which was used to specifically stain nerve fibres and neurons, revealed colocalisation of NF200 with Ifnlr1 in the cochlear nerve fibres and spiral ganglion neurons (figure 2D,E). A magnified imaged of Ifnlr1 expression in the organ of Corti revealed high expression of Ifnlr1 in the cytoplasm and nucleoplasm of most cells, although their distributions in these cells were somewhat variable. In contrast to the expression of Ifnlr1 in cytoplasm and nucleoplasm of IHCs, Ifnlr1 was negative in the nucleoplasm in OHCs (figure 2C,H–J). We observed a similar colocalisation pattern of Ifnlr1 and phalloidin in the crista ampullaris of vestibular epithelium (figure 2F).

Immunofluorescence analysis of Ifnlr1 expression in the inner ear of P30 mice. (A) Schematised localisation of Ifnlr1 in the cochlea and spiral ganglion. Staining appears in IHC, OHC, ISC, IPC, OPC, SC, SP, SG, CN, VM, BM and ESCs. The blank cochlea diagram is from the hereditary hearing loss homepage (http://hereditaryhearingloss.org). (B) Overview of Ifnlr1 expression (green) in the cochlea. (C) Details of the organ of Corti showing that Ifnlr1 is highly expressed in the cytoplasm and nuclei of most cells with the exception of the OHCs, which exhibit Ifnlr1 expression only in cytoplasm and not in the nuclei. (D–E) Details of the cochlear nerve (D) and the spiral ganglion (E), in which the structures are clearly stained by antibody directed against the heavy chain of neurofilament (NF200). The merged picture shows partial overlap of Ifnlr1 and NF200 in the nerve fibres and the SG cells. (F) Details of the crista ampullaris showing staining in the vestibular hair cells, supporting cells, stereocilia and nerve fibres. (G–J) Expression of Ifnlr1 (green) in optical section of whole mount of Corti. (G) Stereocilia level. Staining is present in stereocilia of IHC and OHC. (H) Level of cuticular plate of IHC. Staining is observed at the cytoplasm of OHC and also in cuticular plate of IHC. (I) Level above OHC and IHC nuclei. Staining is observed in IHC and OHC cytoplasm. (J) Level of OHC and IHC nuclei. Staining is observed in IHC nuclei, and OHC nuclei are not stained. Nuclei were visualised with DAPI (blue) and F-actin with phalloidin (red). BM, basilar membrane; CN, cochlear nerve; ESCs, external sulcus cells; IHC, inner hair cells; IPC, inner pillar cells; ISC, inner sulcus cells; OHC, outer hair cells; OPC, outer pillar cells; SC, support cells; SG, spiral ganglion; SP, spiral ligament; VM, vestibular membrane.
Aberrant ifnlr1 function causes late-stage developmental defects
The ifnlr1 knockdown MO exerted no significant change in the early development of zebrafish from 32-hpf to 4-dpf (online supplementary figure 2). Circulation in the intersegmental vessels (ISV) appeared normal in both the control and ifnlr1-e4i4-MO-injected fish (online supplementary movie 1). Knockdown of ifnlr1 did not induce organ-specific apoptosis (online supplementary figure 3) or macrophage migration (online supplementary figure 4), indicating that apoptosis and inflammation might not be the cause of reduced hair cell numbers.
Supplementary file 2
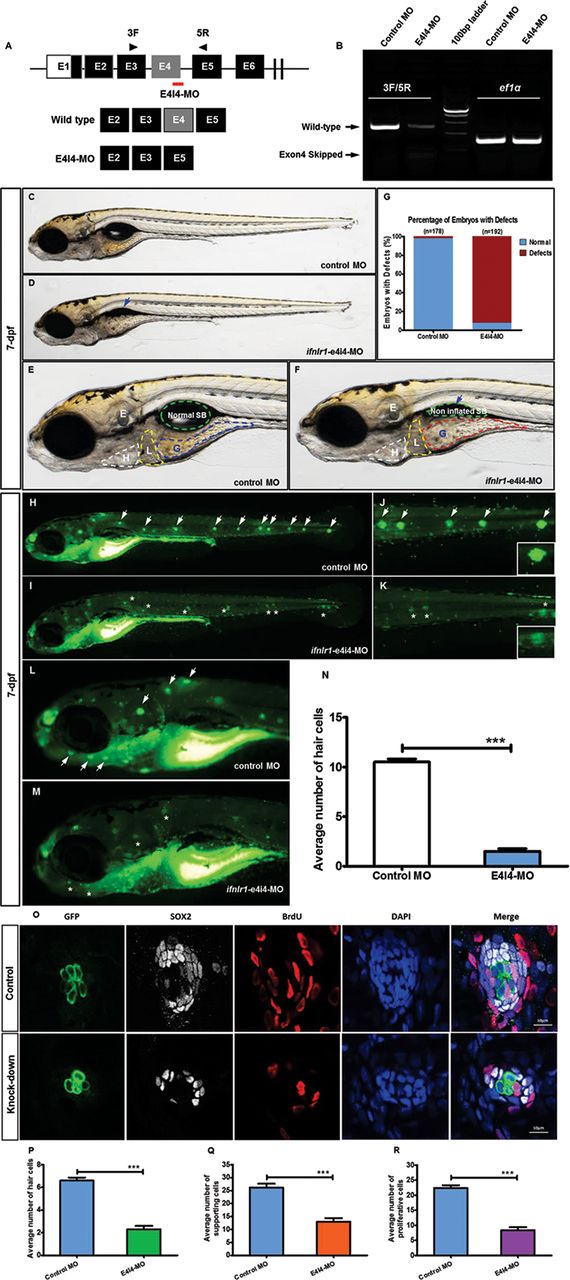
Phenotypes of ifnlr1 zebrafish morphants. (A,B) Effectiveness of ifnlr1 knockdown was confirmed by RT-PCR. (A) The zebrafish ifnlr1 gene was targeted by specific MO antisense to prevent the appropriate splicing of exon 4 (E4I4-MO). Primers 3F and 5R detected the presence of WT (non-mutant) transcripts or those in which exon 4 had been skipped. The diagram below is a schematic depiction of the exon 4-skipped transcript in the E4I4-MO-injected embryos compared with control embryos. (B) RT-PCR of ifnlr1 transcript from control-MO and E4I4-MO MO-injected embryos at 4 dpf demonstrates skipping of exon 4. Injection of 4 ng of ifnlr1 MO altered the splicing between exon 4 and intron 4, as revealed by a shift in PCR bands between control (249 bp) and ifnlr1 MO-injected embryos (67 bp). (C–G) At late stages, knockdown ifnlr1 caused non-inflation of the SB (F, blue arrow) and expansion of the gut lumen area (F, red dashed lines). The bar graph in Panel G shows the percentage of embryos with development defects at 7 dpf. (H–N) ifnlr1 knockdown induces potent neuromasts loss in zebrafish. Control embryos and embryos injected with 4 ng ifnlr1-e4i4-MO were stained with the mitochondrial potentiometric dye DASPEI at 7 dpf. Neuromasts stereotypically located on the lateral line were stained as green dots (H,J: white arrow). Control MO-injected zebrafish had normal numbers of neuromasts. In contrast, significantly decreased hair cell staining (I,K: asterisk) and head neuromast staining (L: white arrow; M: asterisk) were observed in ifnlr1 morphants. Fluorescent DASPEI images were inverted for particle analysis. The fluorescence particle signals of neuromasts were quantified using morphometric analysis. (O) Hair cells, supporting cells and proliferated cells per neuromast were counted and analysed in 6dpf Tg (Brn3c:mGFP). Transgenic zebrafish larvae using four markers GFP (hair cells, green), SOX2 (supporting cells, white), BrdU (proliferated cells, red) and DAPI (nucleus, blue). Four dpf embryos were exposed to BrdU for 48 hours and visualised the cell proliferation. There were fewer hair cells, supporting cells and proliferated cells in morphants than WT. (N,P–R) Quantification of the average number of neuromasts (N), hair cells (P), support cells (Q) and proliferated cells (R) shows significantly decrease in ifnlr1 morphants. Error bars, SEM; ***P<0.0001 (n=10; Student’s t-test). BrdU, bromodeoxyuridine; DASPEI, 2-(4-(dimethylamino)styryl)-N-ethylpyridinium iodide; dpf, days postfertilisation; E, ear; H, heart; L, liver; MO, Morpholino; SB, swim bladder; WT, wild type.

Coinjection of ifnlr1 mRNA from non-mutant zebrafish rescued the phenotypes of non-inflated SB, hair cell loss and head neuromast loss (6 dpf) induced by ifnlr1-e4i4-MO. (A) Lateral views of control MO injected zebrafish embryos. (B) Embryos injected with ifnlr1 MO oligonucleotides (MO), ifnlr1 MO, plus non-mutant zebrafish ifnlr1. (C) Coinjection of non-mutant zebrafish ifnlr1 mRNA rescued non-inflated SB (red arrow) in ifnlr1 morphants at 6 dpf. (D) The bar graph shows the percentage of embryos with developmental defects. (E–N) Defective neuromasts in zebrafish. DASPEI (green) labelled neuromasts at 6 dpf. Neuromasts were stained as green dots (white arrow). The boxed regions are shown at higher magnification (right panels). (E,H,K) Control MO-injected zebrafish had normal numbers of hair cells. (F,I,L) Significantly decreased neuromast staining (asterisk) was observed in ifnlr1 morphants. (G,J,M) Coinjection of ifnlr1 mRNA from non-mutant zebrafish rescued ifnlr1-e4i4-MO from inducing hair cell loss and head neuromast loss (white arrow). Fluorescent DASPEI images were inverted for particle analysis. (N) The fluorescence particle signal in neuromasts was quantified using morphometric analysis. Quantification of the average number of neuromasts in the control embryos and the embryos injected with ifnlr1 MO or ifnlr1 MO plus non-mutant zebrafish ifnlr1. Error bars, SEM; ***P<0.0001 (n=10; ANOVA). DASPEI, 2-(4-(dimethylamino)styryl)-N-ethylpyridinium iodide; MO, Morpholino; SB, swim bladder.
In late stages of development, ifnlr1 knockdown caused non-inflation of the swim bladder and expanded the gut lumen area. The effectiveness of ifnlr1 knockdown was confirmed by RT-PCR.
Disruption of the developmental processes in the inner ear in ifnlr1-morphant zebrafish
Compared with normal controls, morphants had a reduced number of neuromasts (figure 3I,M). Hair cells and supporting cells per neuromast were counted in 6 dpf Tg (Brn3c:mGFP) transgenic zebrafish larvae using four markers, GFP (hair cells), SOX2 (supporting cells), BrdU (proliferated cells) and DAPI (nucleus). Fewer hair cells and supporting cells per neuromast were detected in the neuromasts of lateral line of MO knockdown larvae than in the wild type (WT) (figure 3O). The average number of hair cells per neuromast in6 dpf zebrafish was 2.3±0.3 for morphants and 0.6±0.26 for WT. The average number of supporting cells per neuromast was 13±1.36 for morphants, while 26.2±1.43 for WT. The average number of proliferated cells per neuromast was 8.4±0.97 for morphants, while 22.4±0.88 for WT. To determine whether the loss of hair cells and supporting cells were related to decreased cell proliferation, we exposed the 4 dpf larvae to BrdU for 48 hours and assessed BrdU incorporation by immunolabeling to visualise the cell proliferation in WT and morphants. The number of BrdU-positive cells was much lower in morphants than in WT (figure 3O,R).
Rescue experiments indicate linkage of the ifnlr1 mutation to reduced numbers of hair cells and non-inflation of the swim bladder in zebrafish
Zebrafish ifnlr1 mRNA was coinjected with MO into one-cell-stage embryos to rescue abnormalities caused by MO in zebrafish, and the number of lateral line neuromasts and the swim bladder size were analysed. The results revealed that WT mRNA could largely rescue the ifnlr1-e4i4-MO-induced non-inflation of the swim bladder and loss of hair cells and head neuromasts at six dpf, suggesting that the IFNLR1 mutation caused hearing loss in family JS4842 (figure 4).
The ifnlr1 mutation may contribute to hearing loss through the Janus kinase (Jak)1/signal transducers and activators of transcription (STAT)3 signalling pathway
The mRNA expression of cytokine receptor family member b4 (crfb4 or interleukin (IL)10 receptor 2, il10r2), jak1, tyrosine kinase 2 (tyk2), stat1a, stat2, stat3, stat4, stat5b and irf9, related to the Jak/STAT signalling pathway, was tested by quantitative real-time PCR using cDNA obtained from 4dpf WT and morphants zebrafish. In ifnlr1-morphant zebrafish, Jak1/STAT3 pathway members were upregulated, including stat3, jak1, il10r2 (also called crfb4), stat5 and tyk2 (figure 5). We postulate that upregulation of Jak1/STAT3 signalling is a characteristic event in ifnlr1-morphant zebrafish.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jak1/STAT3 signalling pathway were upregulated in ifnlr1-morphant zebrafish. (A,C) Schematic model illustrating the mechanism of action of ifnlr1 on the Jak1/STAT3 signalling pathway. (B) Endogenous crfb4 (il10r2), jak1, tyk2, stat1a, stat2, stat3, stat4, stat5b and irf9 in control and ifnlr1 morphants assessed by qRT-PCR (n=10–15 individual embryos). ns, not significant.
Discussion
This study identified a mutation in IFNLR1 as a cause of ADNSHL at the DFNA2 locus, a region in which mutations in GJB3 (MIM 603324) and KCNQ4 (MIM 603537) were discovered previously. We focused on this genomic region because a significant linkage was identified in a genome scan, and we found a missense mutation in IFNLR1 via whole-exome sequencing.
A previous study found that IFNLR1 is expressed in a highly tissue-specific manner, with primary expression in the epithelial and liver tissues in humans.16 However, no studies have reported IFNLR1 expression in the auditory system. In this study, we found broad expression of Ifnlr1 in the inner ear, including sensory and non-sensory epithelia of the organ of Corti, cochlear nerve and spiral ganglion of the cochlea and in the crista ampullaris of vestibular organ. To exclude the non-specific reaction of polyclonal antibiotics, we applied two polyclonal antibiotics against mouse Ifnlr1 from different companies (Bioss and Abcam) and the results demonstrated similar Ifnlr1 localisations. In the affected individuals of family JS4842, DPOAE and ABR were absent, excluding the possibility of auditory neuropathy spectrum disorders. Although Ifnlr1 was expressed abundantly in both the cochlea and the vestibulum in mice, the affected individuals of the JS4842 family showed no symptoms of vestibular dysfunction. Despite the wide tissue distribution of IFNLR1, IFNLR1 missense mutation identified in this study only influenced hearing, as additional clinical signs outside the auditory system were absent in the affected individuals. Such an observation is not unusual; non-syndromic hearing loss is associated with mutant alleles of several ubiquitously expressed genes such as ILDR1 (MIM 609739), TPRN (MIM 613354), MARVELD2 (also known as TRIC, MIM 610572), HGF (MIM 142409) and ACTG1 (MIM 102560).17–21 Whether hearing impairment is due to disruption of IFNLR1 function in hair cells, supporting cells, cochlear nerve or all of these remains to be addressed in future studies.
IFNLR1 has seven exons, and its coding protein, IFNLR1, has one functional domain, tissue factor domain (19–111 aa). This domain is similar to the fibronectin type-III domain and is found in cytokine receptors, interleukin and IFN receptors and coagulation factor III proteins.22 23 The missense mutation c.296G>A causes an amino acid substitution from arginine to histidine at position 99, and this mutation is predicted to affect the tissue factor domain of IFNLR1 (figure 3C).
The functional IFN-λs receptor complex is composed of IFNLR1 and IL-10R2 chains.4 5 The specific activity of IFN-λs is determined, in part, by the expression level of its receptor chain IFNLR1, which is expressed primarily in epithelial cells and a specific subsets of immune cells.24–29 IFN-λs appear to be the major IFN type produced by both murine and human airway epithelial cells in response to various respiratory viruses,30–32 and recent studies have suggested the functional importance of IFN-λs beyond innate antiviral protection.
To our knowledge, this is the first report to suggest a role of IFNLR1 in hearing. To mimic the IFNLR1 loss of function, we performed experiments in zebrafish using MO to knockdown ifnlr1. We observed a reduced number of hair cells, supporting cells in the lateral line in morphants compared with WT. Moreover, rescue experiments revealed that WT mRNA could largely rescue MO phenotypes.
At late-stage development in morphant zebrafish (7 dpf), ifnlr1 knockdown caused non-inflation of the swim bladder and expansion of the gut lumen area. The swim bladder in fish is a hydrostatic organ, postulated as a homologue of the tetrapod lung. However, the affected individual of family JS4842 showed no lung disorder symptoms. It is possible that IFNLR1 has a variety of functions depending on the species or tissue distribution. The mechanism of swim bladder inflation in late-stage development of ifnlr1 morphant zebrafish has yet to be elucidated.
IFN-λ-induced receptor engagement leads to the tyrosine phosphorylation of the IFNLR1 intracellular domain via the activation of receptor-associated Jak kinases, Jak1 and Tyk2, and to the subsequent activation of latent transcription factors of the STAT family: STAT1, STAT2, STAT3, STAT4 and STAT5.4 11 33 Increasing evidence points to a role for the Jak/STAT signalling pathway in the auditory system. Inhibition of the Jak2/STAT3 signalling pathway is protective against noise-induced cochlear tissue damage and loss of hearing sensitivity.34 RNA-Seq analysis revealed that the Jak1/STAT3 pathway and cell cycle are activated following zebrafish hair cell death.35 The Jak1/STAT3 pathway is upregulated during zebrafish inner ear regeneration.36
This study identified IFNLR1 as a gene involved in autosomal-dominant hearing impairment. Several lines of evidence support the hypothesis that the identified IFNLR1 mutation is responsible for autosomal-dominant hearing impairment: (1) IFNLR1 c.296G>A completely cosegregated with hearing loss in an ADNSHL family; (2) the identified mutation influenced the conserved residue of the IFNLR1 protein and in silico analysis suggested that this mutation disturbed protein function; (3) the identified mutation was absent in 3396 ethnically matched controls (2896 in-house controls and 500 controls with normal hearing); (4) Ifnlr1 was expressed in the inner ears of mice and (5) ifnlr1 knockdown induced hair cell and supporting cell loss in zebrafish. Further research is needed to address the spectrum and prevalence of IFNLR1 mutations in ADNSHL families of different ethnicities, the various functions of the different IFNLR1 isoforms and possible ligands and interaction partners. Further characterisation of the exact molecular mechanisms by which genetic defects in IFNLR1 cause non-syndromic hearing loss will contribute to our understanding of the essential processes for normal hearing, which might lead to the development of therapeutic interventions.
Acknowledgments
We sincerely thank all the family members for their participation and cooperation in this study. We thank Professor Hua-Wei Li and D. Dong-Mei Tang of Fudan University for sharing the Tg (Brn3c:mGFP) S356T transgenic zebrafish and many supports. We are also grateful to Dr Zhuan-Bin Wu of Shanghai Biomodel Organism Science & Technology Limited and Dr Guang-Chun Bai of Albany Medical College for their valuable comments and suggestions.
References
Footnotes
XG, Y-YY and Q-FL contributed equally.
Contributors XG, Y-YY, Q-FL and PD conceived of the study and participated in its design and drafted the manuscript. J-CX, L-PG, S-SH, H-KL and G-JW participated in the data interpretation and analysis. Y-HQ, W-QW, S-WQ, FX, YS, M-YH, YJ, DB and D-YK participated in the experiment in vivo and in vitro. All authors read and approved the final manuscript.
Funding This work was supported by National Natural Science Foundation of China (81730029, 81371096) and National key research and development project (2016YFC1000700, 2016YFC1000704) to PD; grants from National Natural Science Foundation of China (81570929) to XG; grants from National key research and development project (2016YFC1000706) and National Natural Science Foundation of China (81371098) to Y-YY; grants from National Natural Science Foundation of China (81360159) and National Basic Research Program of China (2014CB541706) to G-JW; grants from Military Health Care Special Project (15BJZ23) to J-CX; grants from National Natural Science Foundation of China (81200751) to S-SH; grants from National Natural Science Foundation of China (81400471) to YS; grants from Natural Science Foundation of Hainan Province (817345) to M-YH.
Disclaimer The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Chinese People’s Liberation Army General Hospital Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.